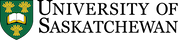
8.6. Reaction: Addition of H-OH
It is possible to add a hydrogen (H) and an alcohol (OH) across an alkene (Scheme 8.8). There are multiple ways of doing this (see Sections 8.6.4. to 8.6.6.) though the most common is to use a strong acid as a catalyst. This is sometimes referred to as hydration.
Scheme 8.8 – Generalized Reaction Equation for Acid-Catalyzed Addition of H-OH Across an Alkene.
8.6.1. Mechanism and Rate-Determining Step
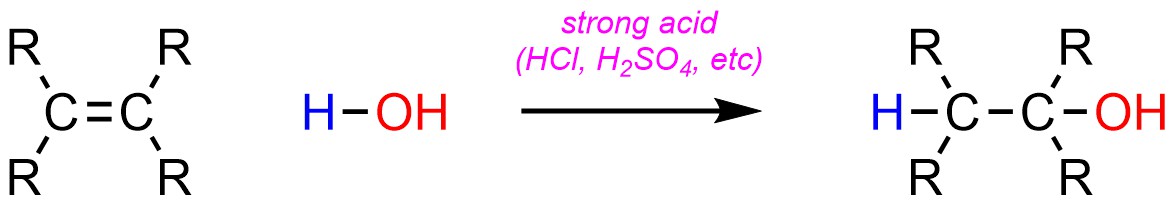
The mechanism for this reaction has the same two main steps but has additional activation and regeneration steps for the catalyst (Scheme 8.9). The strong acid reacts with water to form hydronium (not shown). This is the active catalyst. First, the electrophile (H2O) gets activated by the catalyst. This greatly increases its electrophilicity. Second, the alkene (nucleophile) attacks hydronium (electrophile). This creates a new C-H bond, a carbocation, and water. Third, the water (nucleophile) attacks the carbocation (electrophile). This creates a new C-O bond. Finally, another equivalent of water removes a proton, generating the final product and regenerating the catalyst.
Scheme 8.9 – Reaction Mechanism for Acid-Catalyzed Addition of H-OH Across the Alkene of (E)-But-2-ene.
As before, carbocation formation requires a large amount of energy and is the rate-determining step. The overall reaction is exergonic.
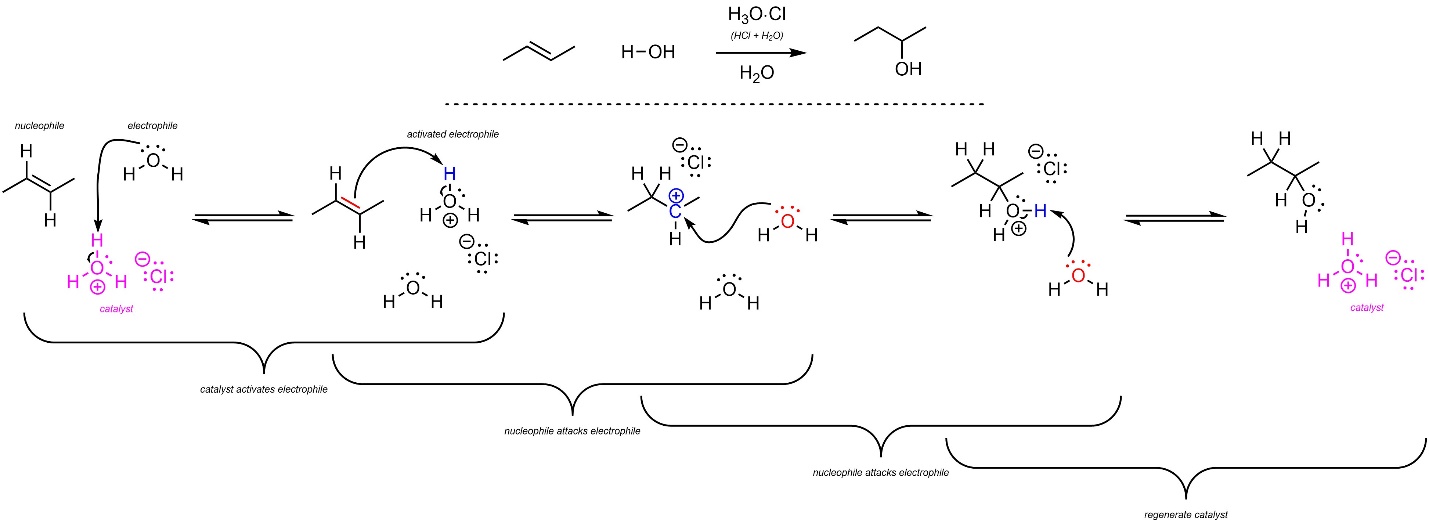
8.6.2. Regioselectivity – Markovnikov Selectivity
As before, the major product will be the one that results from the more stable carbocation intermediate (the Markovnikov product; Scheme 8.10).
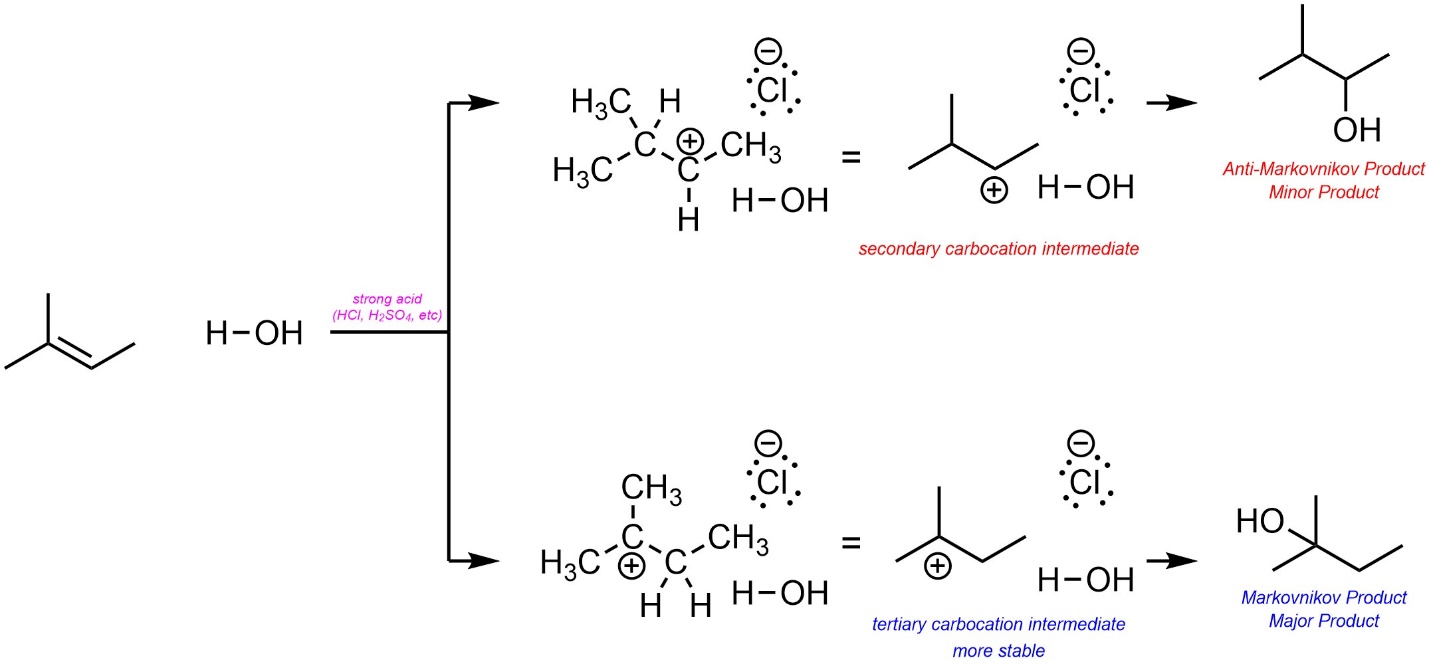
Scheme 8.10 – Determination of Major Product (Regioisomer) for Acid-Catalyzed Addition of H-OH Across the Alkene of 2-Methylbut-2-ene.
This could also be demonstrated using overlapped reaction coordinates for the two pathways.
8.6.3. Stereoselectivity
All stereoselectivity considerations for this reaction are identical to those for the addition of H-X across an alkene (see Section 8.5.3.). Assuming there are no pre-existing stereocentres in the starting material, the first non-catalysis step (alkene attacks hydrogen) will not be stereoselective. If a new stereocentre is formed during this step, it will be formed as an equal mixture of both (R) and (S). Assuming there are no stereocentres in the intermediate, the second non-catalysis step (water attacks carbocation) will not be stereoselective. If a new stereocentre is formed during this step, it will be formed as an equal mixture of both (R) and (S). In either step, if another stereocentre (or multiple stereocentres) exists then the resulting intermediates/products will be diastereomers. The two stereoisomers are NOT formed equally and the product is a non-one-to-one mixture of diastereomers. In these cases, it is important only to recognize that the product will be formed as a mixture of diastereomers. Predicting which diastereomer should be favoured is not required.
8.6.4. Variant: Addition of H-OR
Instead of H-OH, it is possible to add a hydrogen (H) and an ether (OR) across an alkene (Scheme 8.11). This reaction is identical to the acid-catalyzed addition of H-OH but uses an alcohol instead of water as the solvent.
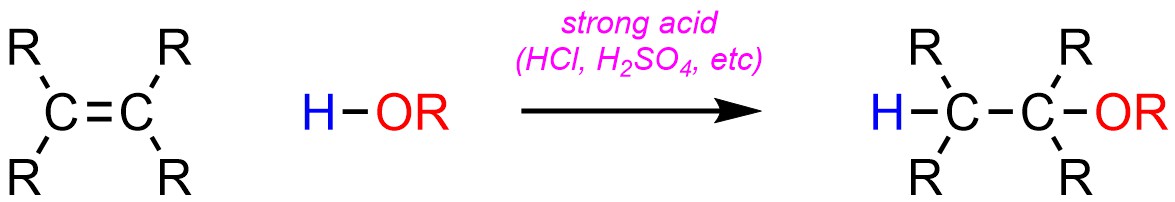
Scheme 8.11 – Generalized Reaction Equation for Acid-Catalyzed Addition of H-OR Across an Alkene.
The mechanism for this reaction is identical to the acid-catalyzed addition of H-OH but uses an alcohol instead of water as the solvent (Scheme 8.12).
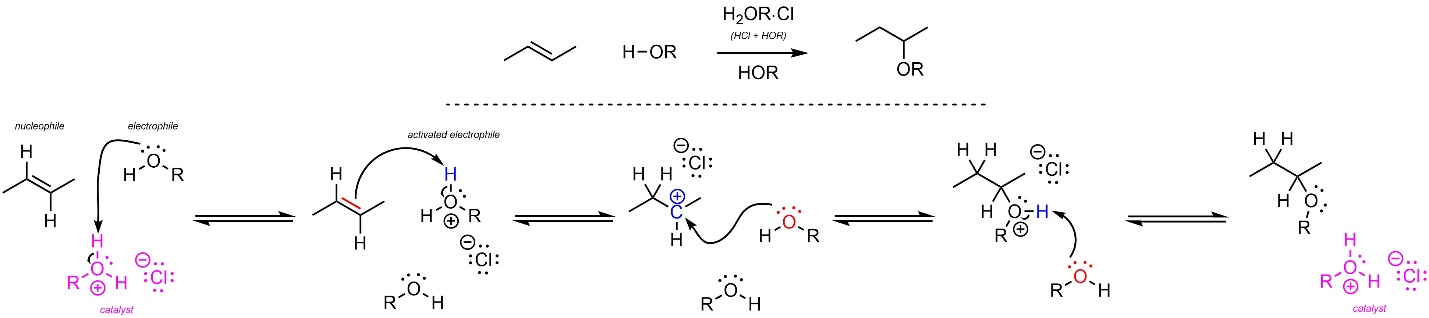
Scheme 8.12 – Reaction Mechanism for Acid-Catalyzed Addition of H-OR Across the Alkene of (E)-But-2-ene.
As before, carbocation formation requires a large amount of energy and is the rate-determining step. The overall reaction is exergonic. As before, the major product will be the one that results from the more stable carbocation intermediate (the Markovnikov product). As before, all stereoselectivity considerations for this reaction are identical to those for the addition of H-X across an alkene.
8.6.5. Variant: Oxymercuration
Using strong acids to add a hydrogen (H) and an alcohol (OH) across an alkene is sometimes problematic. For example, a molecule may have other functional groups that are destroyed (decomposed) by strong acids. In these cases Oxymercuration-Demercuration may be used instead (Scheme 8.13). The overall reaction requires two steps, one to attach the mercury and alcohol, and one to remove the mercury. It is important to recognize that this is not catalysis; the mercury is not a catalyst.
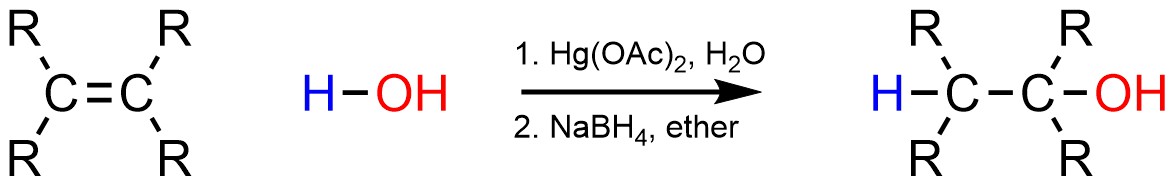
Scheme 8.13 – Generalized Reaction Equation for Oxymercuration-Demercuration Addition of H-OH Across an Alkene.
The mechanism for this reaction is different from the acid-catalyzed addition of H-OH (Scheme 8.14). First, the mercury reacts with the alkene to create a three-membered ring. This step is equivalent to the alkene (nucleophile) attacking the mercury (electrophile) at the same time that the mercury (nucleophile) attacks the “carbocation” (electrophile). Mercury is able to do both steps at the same time, avoiding the formation of an actual high-energy carbocation. Water (nucleophile) then attacks the carbon (electrophile). Although the carbon is not a carbocation, the C-Hg bond is both weak and polar due to the cationic charge on mercury. Acetate then removes the extra proton and generate the oxymercuration product. The mechanism for the demercuration step is slightly complex and is generally omitted from introductory material.
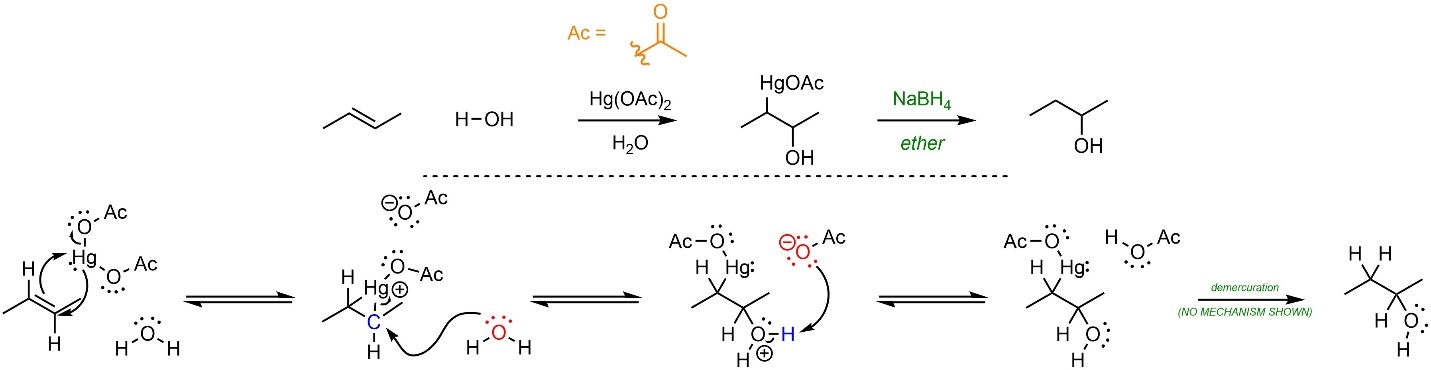
Scheme 8.14 – Reaction Mechanism for Oxymercuration-Demercuration Addition of H-OH Across the Alkene of (E)-But-2-ene.
Because no carbocation is formed, the rate-determining step is now the nucleophilic attack of water onto the carbon. The overall reaction is exergonic.
As before, the major regioisomer will be the Markovnikov product. This is due to the development of a partial cationic charge on the carbon atom during the transition state, which has a similar effect to an energy difference in carbocation intermediates (Scheme 8.15). At an introductory level drawing transition states would not be expected, but students should be able to recognize and compare transition states if they are provided.
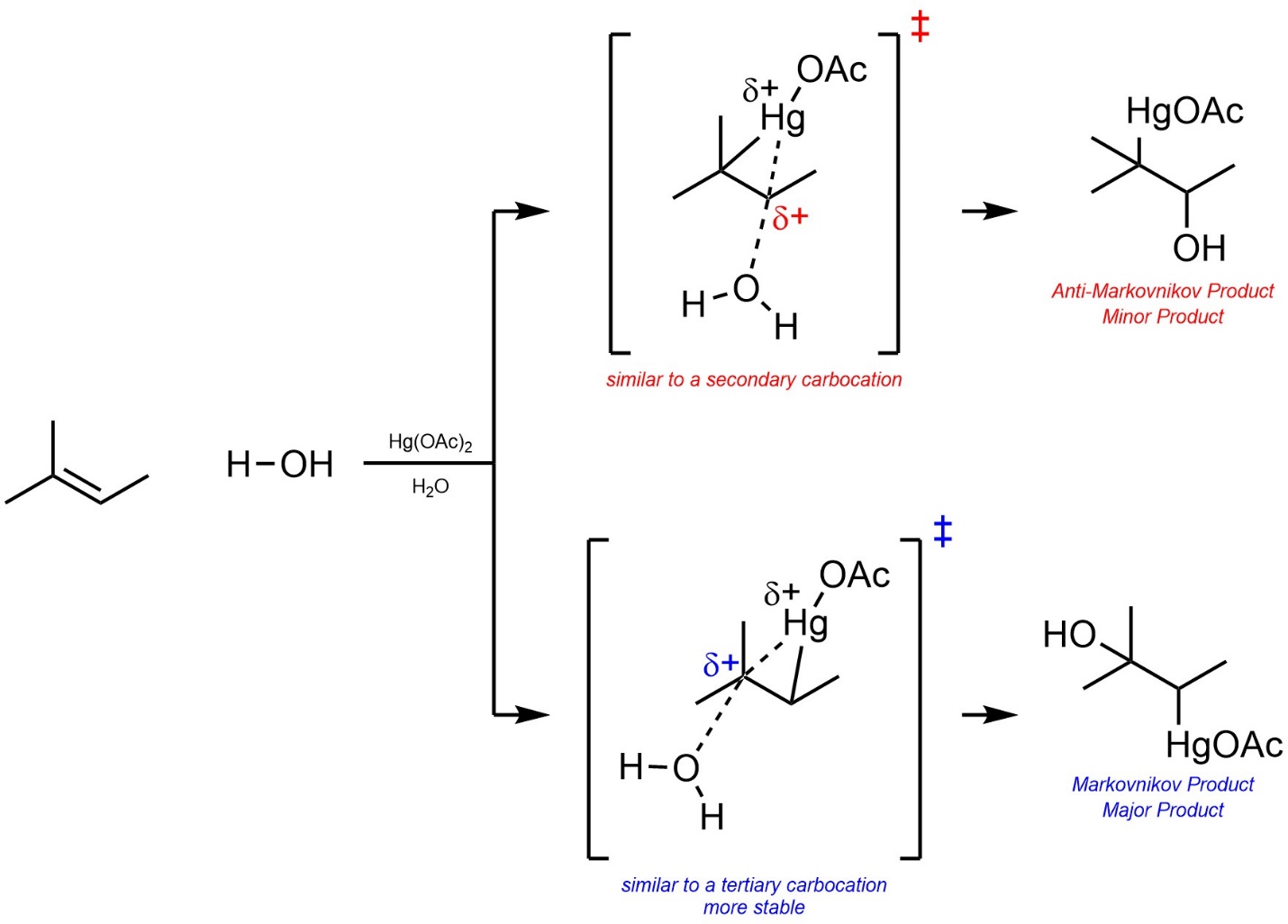
Scheme 8.15 – Determination of Major Product (Regioisomer) for Oxymercuration of the Alkene of 2-Methylbut-2-ene.
Stereoselectivity in oxymercuration-demercuration reactions is possible but challenging to discuss without knowledge of how the demercuration step occurs. Unless otherwise indicated, assume stereoselectivity is not important for these reactions.
8.6.6. Variant: Hydroboration
Acid catalysis and oxymercuration-demercuration both add H-OH across an alkene and are regioselective for the Markovnikov product. An alternative approach, hydroboration (sometimes called hydroboration-oxidation), selectively forms the other regioisomer (the Anti-Markovnikov product; Scheme 8.16). The overall reaction requires two steps, one to attach the boron and hydrogen, and one to replace the boron with an alcohol. It is important to recognize that this is not catalysis; the boron is not a catalyst.
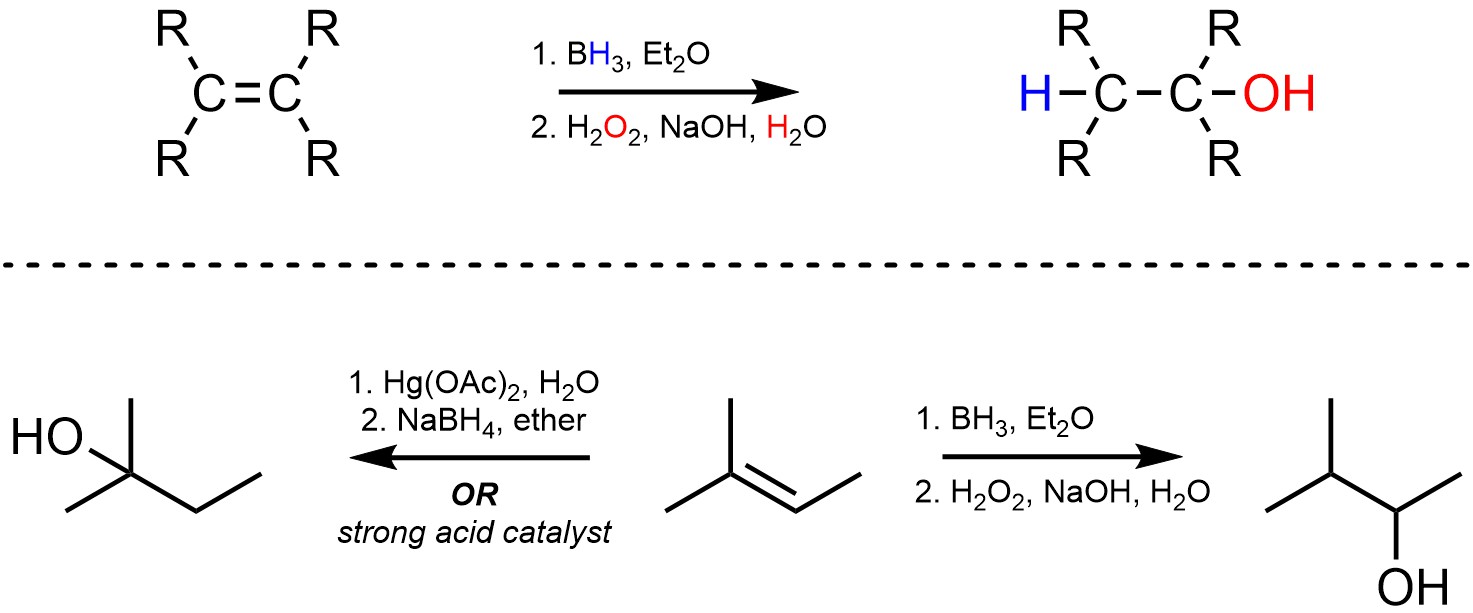
Scheme 8.16 – Generalized Reaction Equation for Hydroboration-Oxidation Addition of H-OH Across an Alkene and Comparison of Regioselectivity with Other Methods.
Recall that B-H bonds are polar, with hydrogen being more electronegative than boron (see Figure 7.5). As with borohydride, the hydrogen atoms have excess electron density and are nucleophilic. Boron is also unusual in that it is semi-stable with an incomplete octet. For example, the boron in borane (BH3) has only 6 electrons in its valence shell but is stable when in ether-containing solvents. However, the incomplete octet means that the boron in borane is (highly) electrophilic.
The mechanism for this reaction is different from both the acid-catalyzed addition of H-OH and oxymercuration-demercuration (Scheme 8.17). Because the hydrogen (nucleophile) is covalently attached to the boron (electrophile), the alkene both attacks the boron and is attacked by the hydrogen at the same time. This avoids the formation of an actual high-energy carbocation and forces a stereospecific addition (see below). An added benefit of this reaction is that the product, an alkylborane (BH2R), has the same reactivity as borane. This means that each equivalent of borane can deliver three hydrogens to three alkenes. The mechanism for the oxidation step is complex and generally omitted from introductory material.
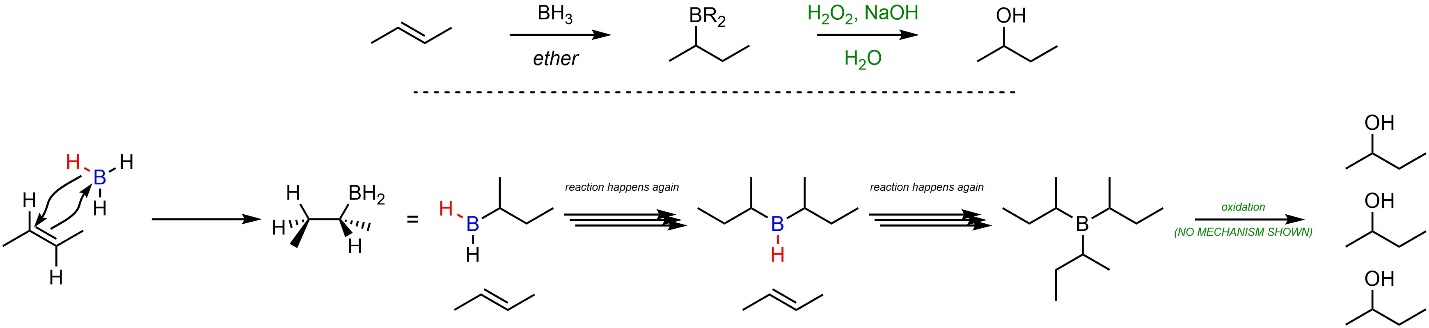
Scheme 8.17 – Reaction Mechanism for Hydroboration-Oxidation Addition of H-OH Across the Alkene of (E)-But-2-ene.
There is only one step in the first reaction. By definition, it is the rate-determining step. The overall reaction is exergonic.
The major regioisomer is the Anti-Markovnikov product. Two factors lead to this. First, the development of a partial cationic charge on the carbon atom during the transition state has a similar effect to an energy difference in carbocation intermediates (Scheme 8.18).
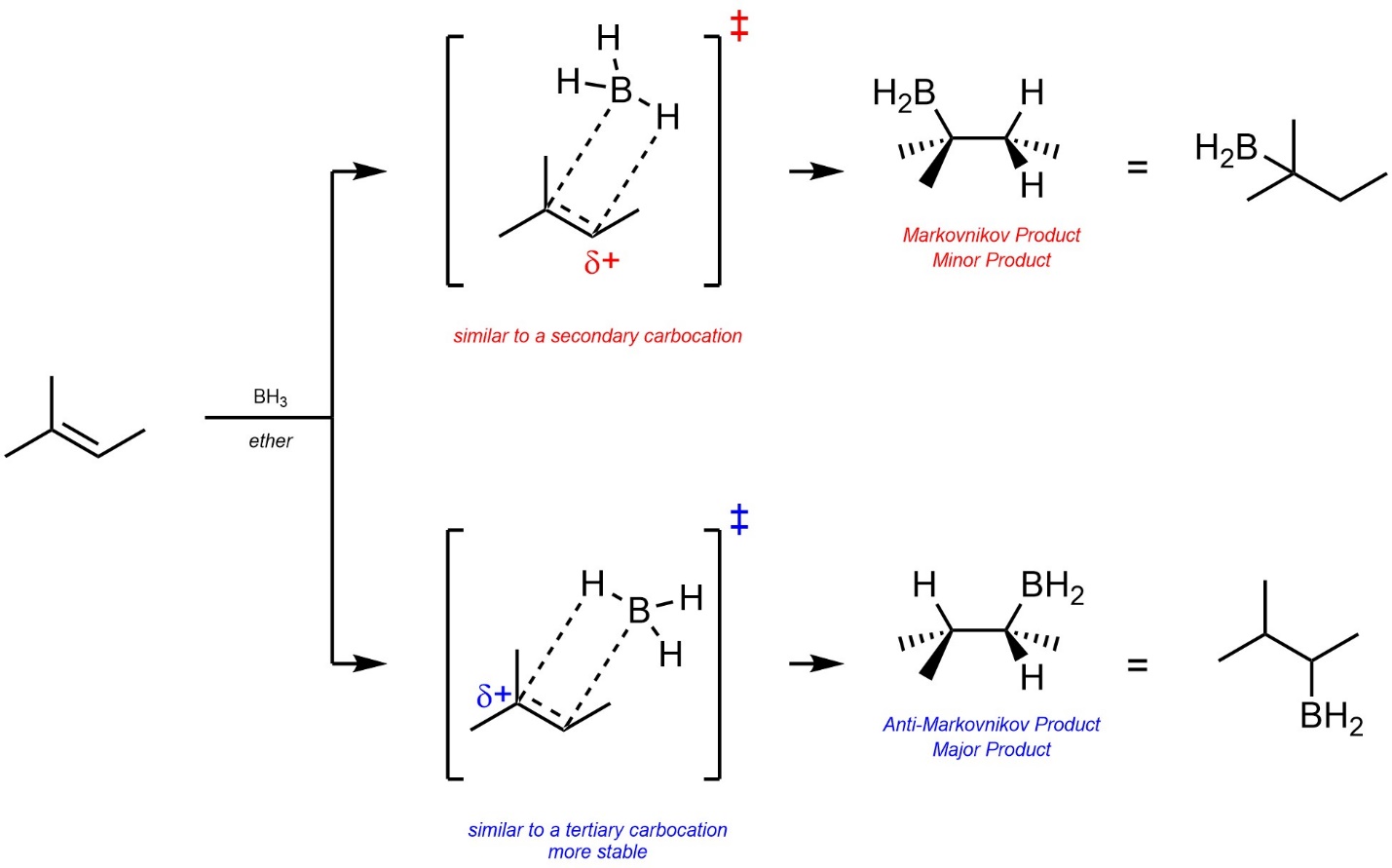
Scheme 8.18 – Partial Cationic Charges During the Transition States for Determination of Major Product (Regioisomer) for Hydroboration-Oxidation of the Alkene of 2-Methylbut-2-ene.
Second, there is steric strain during the first addition (BH3 + alkene) between the hydrogens of the borane and the hydrogens of the alkyl substituents of the alkene (Scheme 8.19). This increases for subsequent additions (BH2R + alkene; BHR2 + alkene), making the regioselectivity even higher. Remember that the B-H bond is located directly above or below the C-C bond of the alkene, it is simply hard to show this in two dimensions.
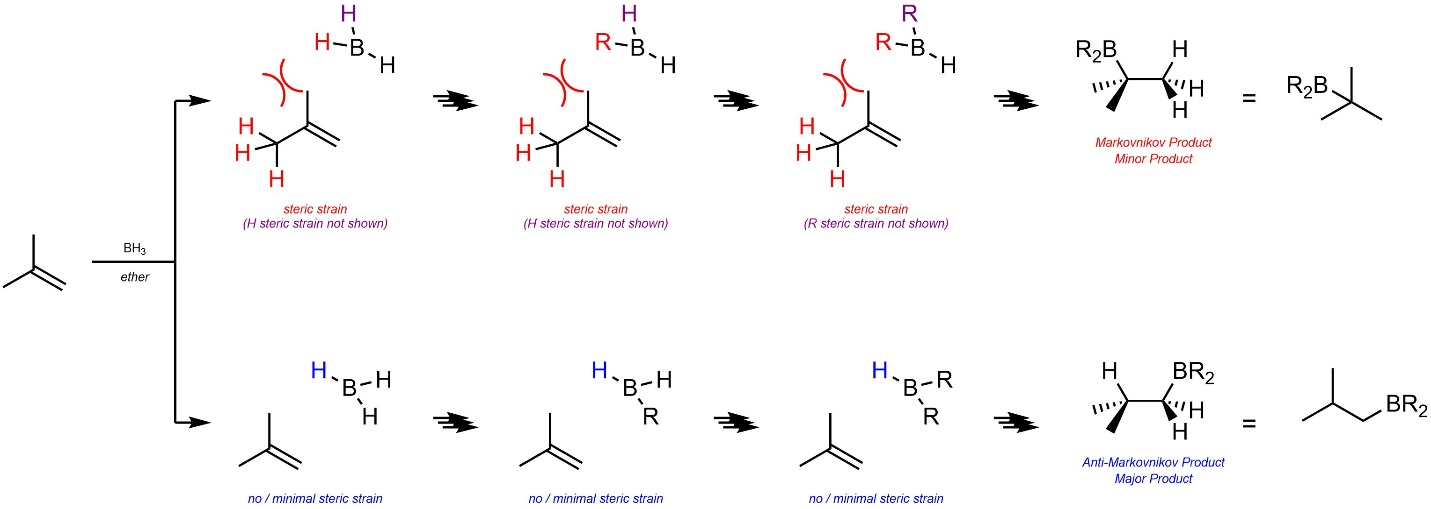
Scheme 8.19 – Steric Strains for Determination of Major Product (Regioisomer) for Hydroboration-Oxidation of the Alkene of 2-Methylprop-1-ene.
Hydroboration-Oxidation reactions are stereospecific (diastereospecific). Because the C-H and C-B bonds are formed at the same time it is not geometrically possible for them to form on different sides (top/bottom) of the alkene; they must both come from above or below the alkene (Scheme 8.20). This is called cis addition, as the two new substituents must be added cis to each other (older sources may refer to this as syn addition). Although the mechanism for the oxidation step is too complex to explore, it is useful to know that the C-B bond is replaced with a C-O bond with retention of configuration; if the C-B bond was “up” (wedged), the C-O bond will also be “up” (wedged) and vice versa.
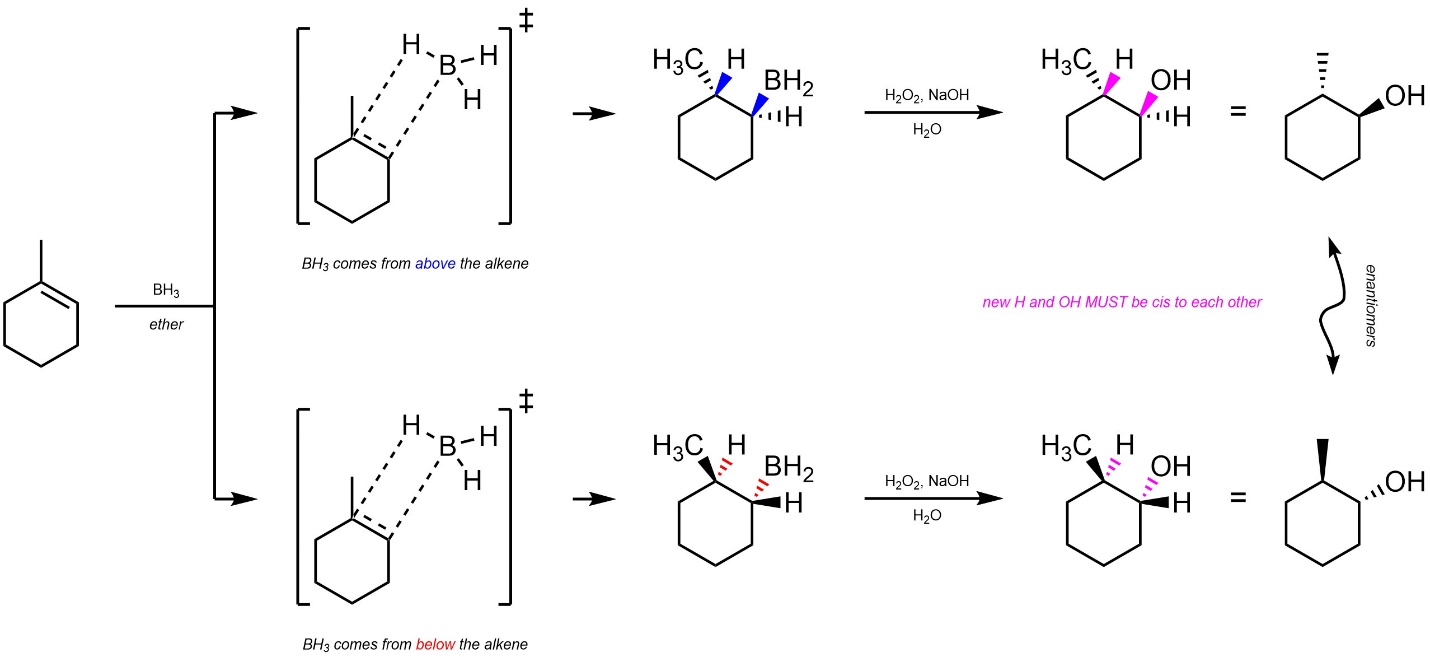
Scheme 8.20 – Diastereospecific Addition of H-OH Across the Alkene in Hydroboration-Oxidation of the Alkene of Methylcyclohexene.
It is very important to remember that this is a form of diastereoselectivity (by being diastereospecific). There is no enantioselectivity.
