8.4. Stereoselectivity
Adding two new groups across an alkene introduces another new complication. To simplify the number of products, consider a cyclic alkene adding the generalized X-X across the double bond (Scheme 8.3). There are different compounds possible as products.
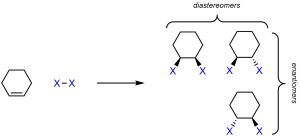
Scheme 8.3 – Stereoisomers from Electrophilic Addition of X-X to Cyclohexene.
8.4.1. Terminology: Stereoselective, Enantioselective, Diastereoselective
For some reactions multiple stereoisomers are possible products. If the stereoisomers are not formed in equal amounts, the reaction is described as being stereoselective (selectively forming more of one stereoisomer over the other(s)).
Stereoselectivity is extremely complex. At an introductory level only an understanding of the basics is required.
Stereoselectivity can be subdivided into two categories. If multiple diastereomers are formed and the reaction favours formation of one diastereomer over the other(s) the reaction is said to be diastereoselective. A complete understanding of how diastereoselectivity can be controlled or achieved lies beyond the scope of this text. Instead, a brief explanation is sufficient to provide context.
Recall that diastereomers are physically and chemically different. As a result, they have different energies. Diastereoselectivity can be achieved if the reaction is fully reversible and there is a moderate to large energy difference between the products (the diastereomers). This is usually referred to as thermodynamic control because the control comes from being able to reach thermodynamic equilibrium. This type of diastereoselectivity is not discussed in this text.
Diastereoselectivity can be achieved if there is a moderate to large energy difference between transition states leading to the products. This is usually referred to as kinetic control because the control comes from one pathway being kinetically faster than the other(s). At an introductory level being able to determine which transition state is higher/lower in energy is functionally impossible without very specific and obvious interactions or geometric requirements. As such, this text will clearly indicate any factor which leads to diastereoselectivity.
Stereoselectivity can be subdivided into two categories. If enantiomers are formed and the reaction favours formation of one enantiomer over the other the reaction is said to be enantioselective. Recall that enantiomers are physically and chemically identical. As a result, they have identical energies. This applies to both products and transition states that are enantiomers of each other. Because of this enantioselectivity is typically much harder to achieve and generally requires advanced reactions, mechanisms, and/or catalysis. No reactions discussed in this text will be enantioselective.
8.4.2. Stereoselective vs. Stereospecific
A reaction is stereoselective if multiple stereoisomers are possible products and one is formed in higher amounts than the other(s). For example, the addition of a nucleophile to (S)-3-iodobutan-2-one (Figure 7.18) results in two possible diastereomers. There are different steric interactions during attacks from the top or bottom sides. As a result, the transition states leading to the two products will have different energies (kinetic control) and more of one diastereomer will be formed than the other. At an introductory level knowing which diastereomer is favoured (top or bottom) and/or by how much (i.e. 60:40 or 99:1?) is not possible.
A reaction is stereospecific if, as a result of the mechanism, only one stereoisomer may be formed. For example, the addition of Br-Br to cyclohexene (Scheme 8.4; see Section 8.7) is stereospecific (diastereospecific). Because of the mechanism it is not physically possible to make ANY of the other diastereomer. The reaction is specific, with 0% of the product being the other diastereomer. For reactions like this, at an introductory level knowing which diastereomer is formed is possible.
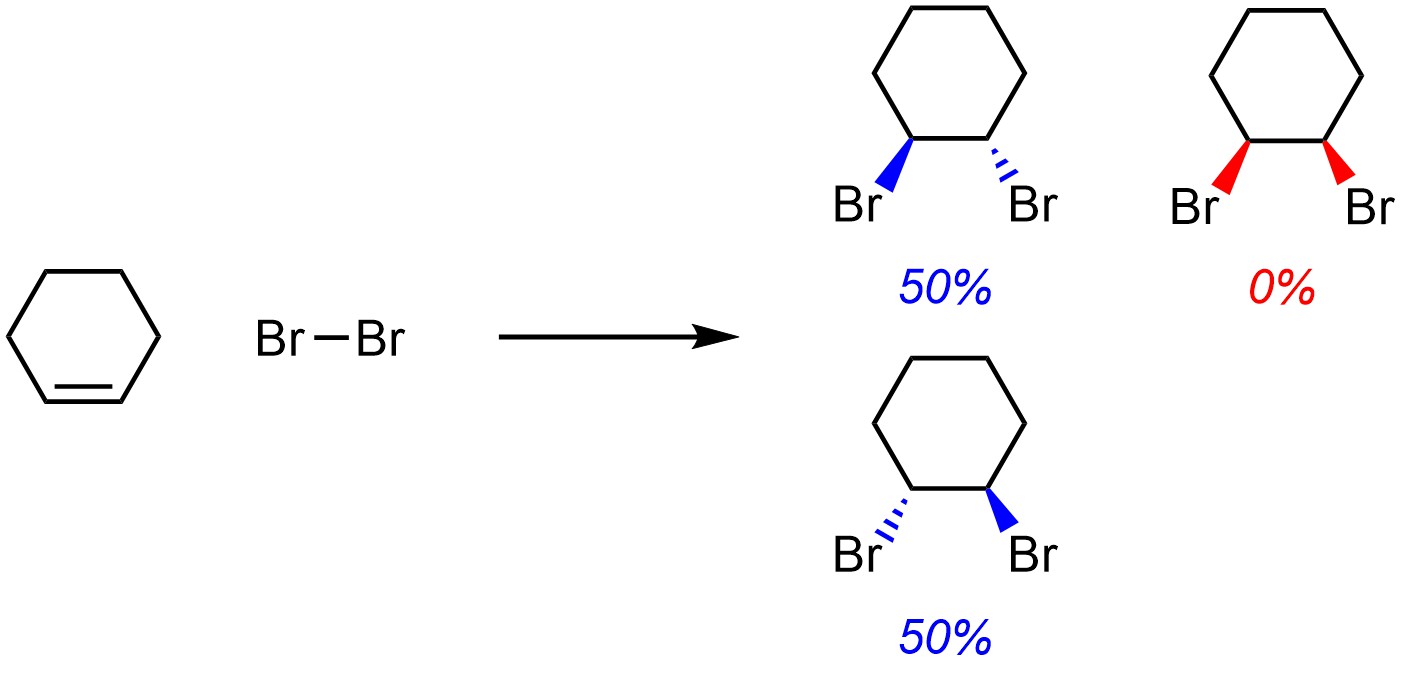
Scheme 8.4 – Example of a Stereospecific (Diastereospecific) Reaction.
Unfortunately, many sources use the term stereospecific (incorrectly) for reactions with high stereoselectivity. This is especially common when discussing enzymatic reactions, where stereoselectivity may be exceptionally high (i.e. >99.999%). However, since it is possible to form the other stereoisomer and since it is the enzyme’s structure, not the mechanism, which is leading to selectivity these reactions are not properly described as stereospecific.
