8.5. Reaction: Addition of H-X
It is possible to add a hydrogen (H) and a halogen (X; X = F, Cl, Br, I) across an alkene (Scheme 8.5). Some sources may refer to this as hydrohalogenation, though this name is not commonly used.
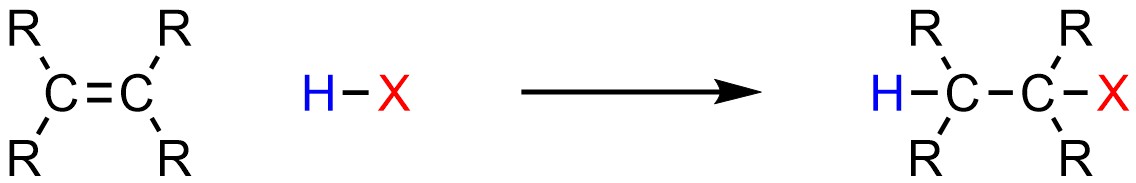
Scheme 8.5 – Generalized Reaction Equation for Addition of H-X Across an Alkene.
8.5.1. Mechanism and Rate-Determining Step
The mechanism for this reaction has two steps (Scheme 8.6). First, the alkene (nucleophile) attacks the hydrogen (electrophile). This creates a new C-H bond, a carbocation, and a halide (anionic halogen). Second, the halide (nucleophile) attacks the carbocation (electrophile). This creates a new C-X bond and generates the final product. This reaction is typically performed in a polar aprotic solvent.
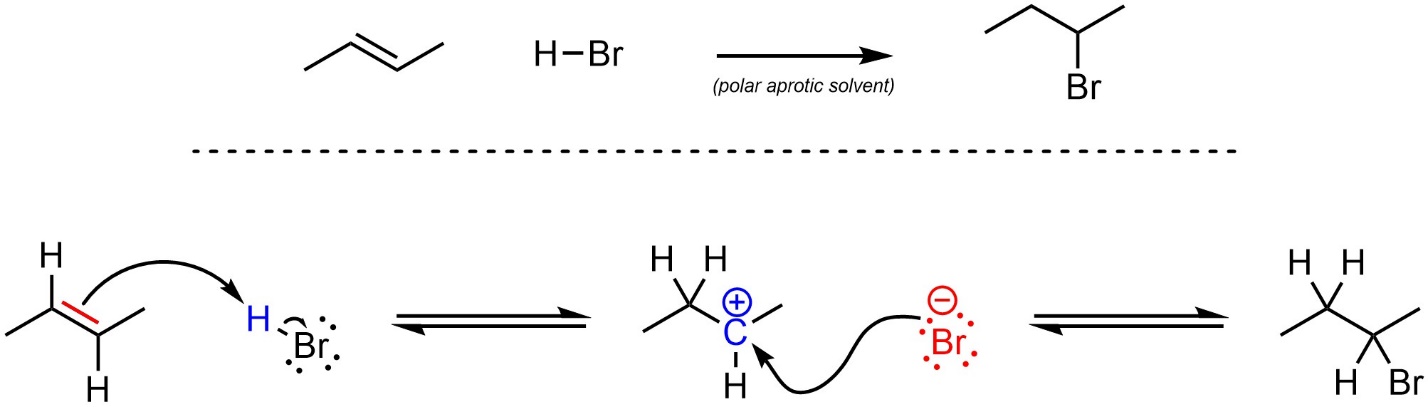
Scheme 8.6 – Reaction Mechanism for Addition of H-Br Across the Alkene of (E)-But-2-ene.
The first step, carbocation formation, requires a large amount of energy and is the rate-determining step (Figure 8.3). The overall reaction is exergonic.
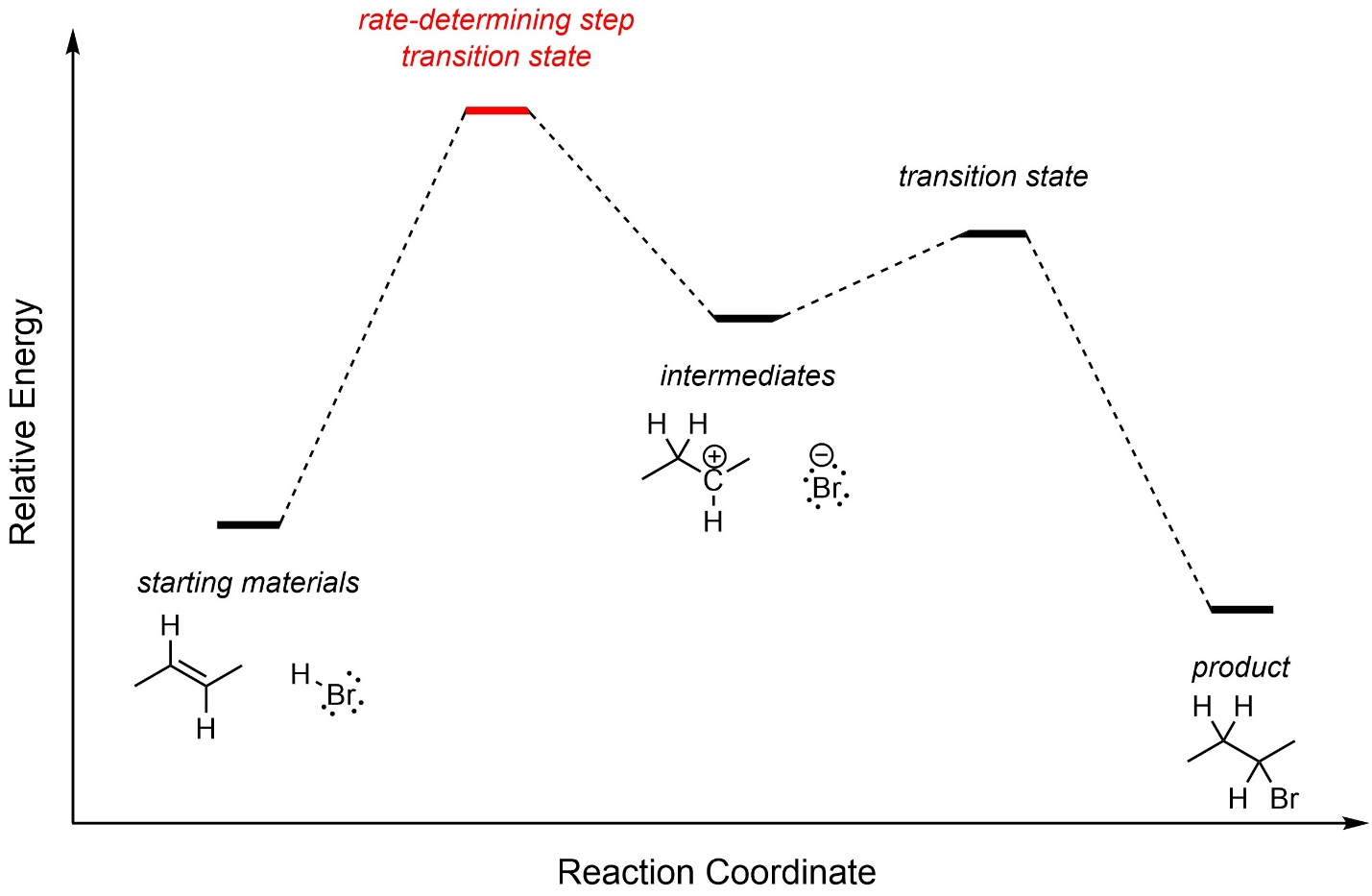
Figure 8.3 – Reaction Coordinate for Addition of H-Br Across the Alkene of (E)-But-2-ene.
8.5.2. Regioselectivity – Markovnikov Selectivity
The rate-determining step involves the formation of a high-energy carbocation intermediate. Usually the more stable an intermediate, the less energy it takes to make the intermediate. When multiple regioisomers are possible in this reaction, they arise from different carbocation intermediates. As a result, the relative stability of carbocations is the main factor leading to regioselectivity.
Carbocations, like other cations, can be stabilized by having resonance delocalization (see Section 6.3). This is the most important factor that can stabilize a carbocation. However, often neither potential carbocation will have multiple resonance forms and instead other factors must be compared.
Recall that having adjacent alkyl groups stabilizes cationic charges (it has no effect on anionic charges). The more adjacent alkyl groups there are, the more stable the cationic charge becomes. Carbocations with fewer alkyl groups adjacent to the cationic atom will be less stable/higher in energy (Figure 8.4). Lacking other factors such as delocalization, tertiary carbocations will be more stable than secondary carbocations, both of which are significantly more stable than primary carbocations. Why this is true is due to hyperconjugation. However, an in-depth discussion of hyperconjugation is too advanced for introductory material.
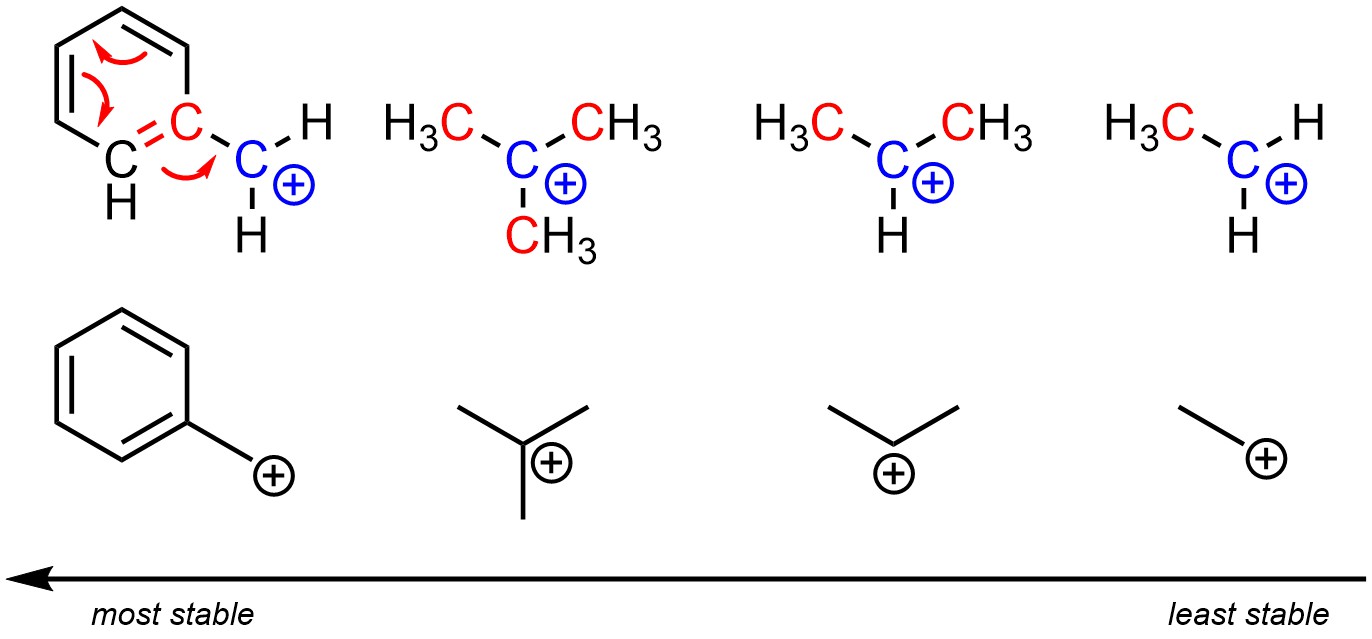
Figure 8.4 – Relative Stability of Carbocations.
Based on this, the major product will be the one that results from the more stable carbocation intermediate (Scheme 8.7). This kind of regioselectivity is sometimes called Markovnikov Selectivity, and the product described as the Markovnikov product. Confusingly, the other regioisomer is sometimes referred to as the Anti-Markovnikov product. The prefix “Anti” here just means “opposite”, there is no relationship to anti conformations (i.e. antiperiplanar, etc.).
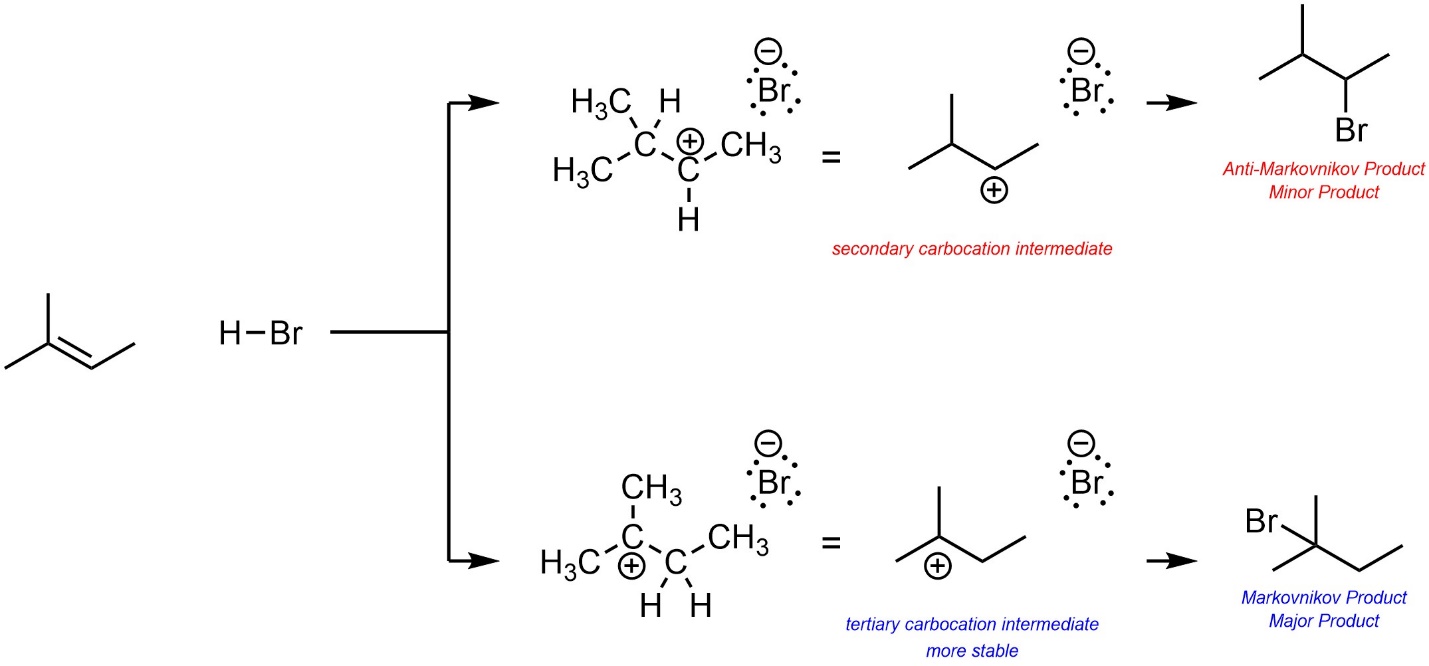
Scheme 8.7 – Determination of Major Product (Regioisomer) for Addition of H-Br Across the Alkene of 2-Methylbut-2-ene.
This can also be demonstrated using overlapped reaction coordinates for the two pathways (Figure 8.5). It should be mentioned that although the two products have slightly different energies, their relative positions on the diagram are irrelevant to the regioselectivity. These reactions are kinetically controlled; only the relative energies of the rate-determining transition states has any effect.
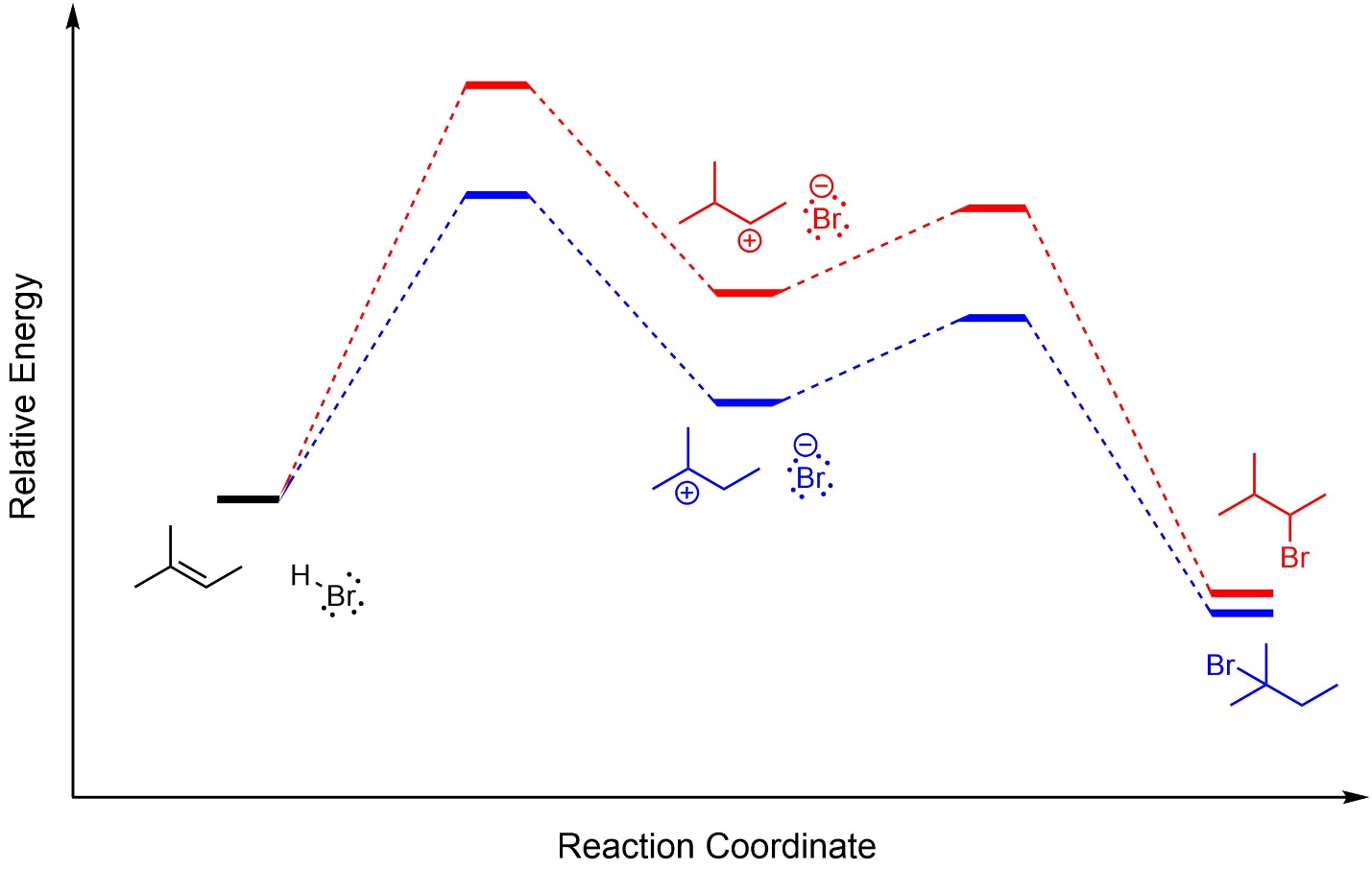
Figure 8.5 – Overlapped Reaction Coordinates for Markovnikov and Anti-Markovnikov Addition of H-Br Across the Alkene of 2-Methylbut-2-ene.
8.5.3. Stereoselectivity
The carbons in alkenes are sp2 hybridized and trigonal planar. The p orbitals which make up the nucleophilic π bond are located both above and below the alkene. Assuming there are no pre-existing stereocentres in the starting material, the first step (alkene attacks hydrogen) will not be stereoselective as attacking from the top of the alkene is sterically and electronically equivalent to attacking from the bottom of the alkene (Figure 8.6). If a new stereocentre is formed during this step, it will be formed as an equal mixture of both (R) and (S). This step is not stereoselective.
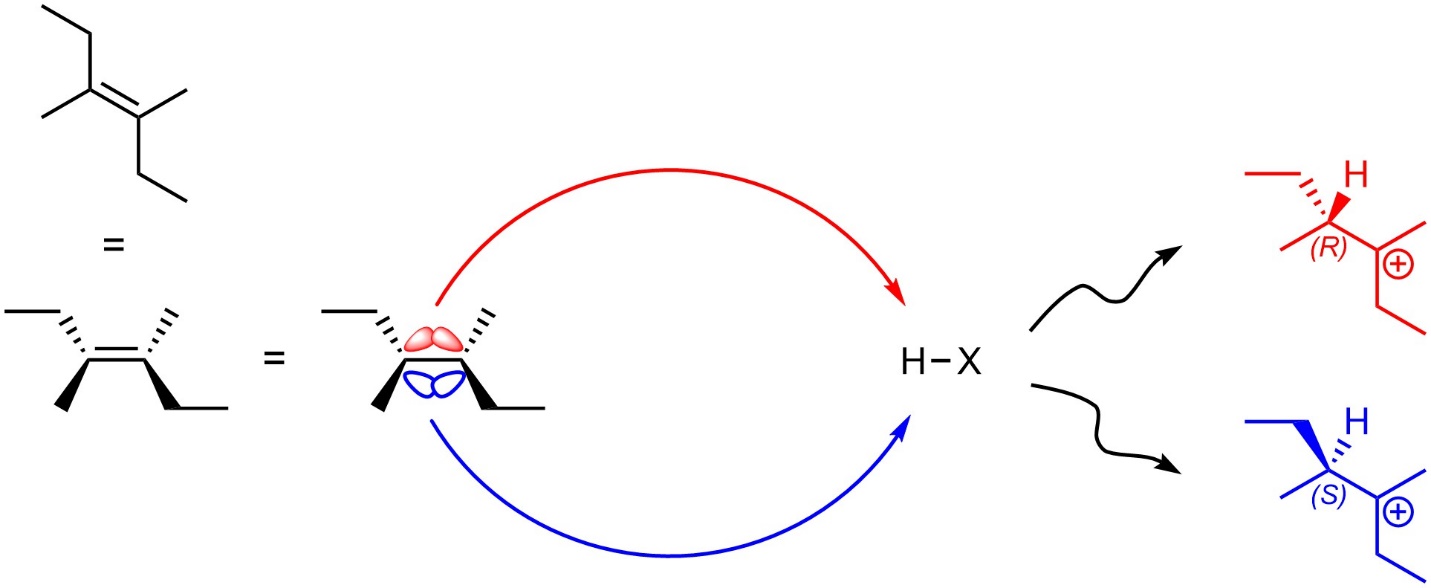
Figure 8.6 – Comparison of Intermediates from Nucleophilic Attacks from the Top and Bottom Sides of 3,4-Dimethylhex-3-ene.
Carbocations are sp2 hybridized, with the p orbital being unoccupied. The empty p orbital which makes up the electrophilic site is located both above and below the carbon. Assuming there are no stereocentres in the intermediate, the second step (halide attacks carbocation) will not be stereoselective as attacking the top of the carbocation is sterically and electronically equivalent to attacking the bottom of the carbocation (Figure 8.7). If a new stereocentre is formed during this step, it will be formed as an equal mixture of both (R) and (S). This step is not stereoselective.
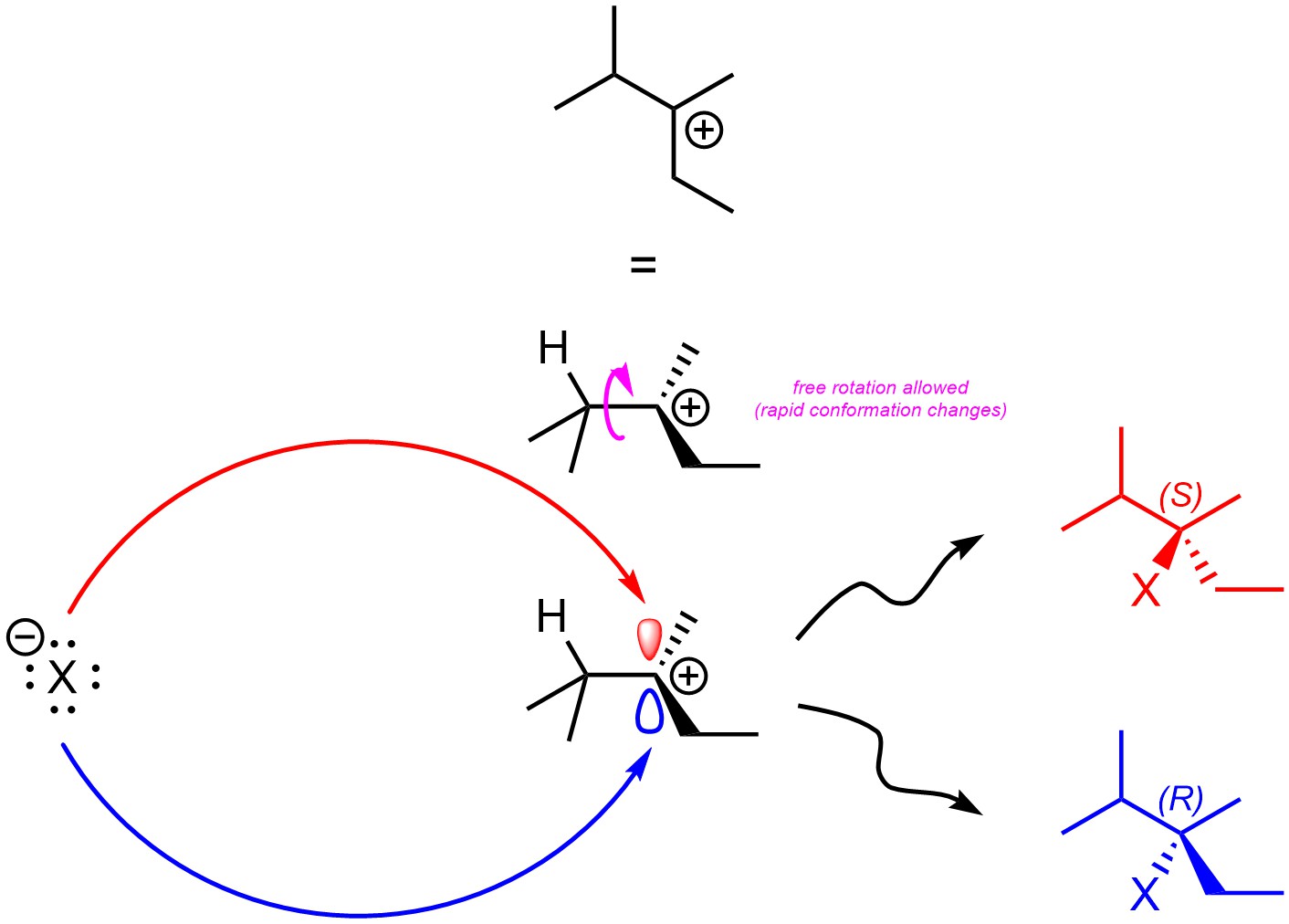
Figure 8.7 – Comparison of Products from Nucleophilic Attacks on the Top and Bottom Sides of 2,3-Dimethylpentan-3-ylium.
In either step, if another stereocentre (or multiple stereocentres) exists then the resulting intermediates/products will be diastereomers. As with nucleophilic attacks on carbonyls (see Section 7.8) because there is a steric and/or electronic difference between attacking the top or bottom in these cases, the two stereoisomers are NOT formed equally and the product is a non-one-to-one mixture of diastereomers. In these cases, it is important only to recognize that the product will be formed as a mixture of diastereomers. Predicting which diastereomer should be favoured is not required.
