Creuser encore davantage
14 Minéralogie du sol
Jim Warren and Graeme Spiers
OBJECTIFS D’APPRENTISSAGE
À la fin de ce chapitre, les étudiants devraient être capables de :
- Identifier les phases minérales les plus communément trouvées dans le sol
- Expliquer la différence entre les minéraux primaires et secondaires
- Décrire l’influence de la glaciation continentale sur la distribution des minéraux dans les sols canadiens
- Décrire la relation entre la taille des particules et les minéraux du sol
- Définir la substitution isomorphe et la base de la capacité d’échange cationique
- Expliquer les différences dans la composition en phyllosilicates des sols entre l’est et l’ouest du Canada
INTRODUCTION
La minéralogie du sol « se rapporte aux minéraux inorganiques trouvés dans la pédosphère et jusqu’à la profondeur où se produit l’altération » (Finkl, 1983). Bien que la minéralogie des sols s’appuie fortement sur les disciplines de la minéralogie, de la géologie, de la chimie inorganique et de la cristallographie, qui sont des disciplines scientifiques en elles-mêmes, le lecteur n’a pas besoin d’avoir de connaissances préalables dans ces domaines pour comprendre les informations présentées dans ce chapitre. L’approche générale adoptée dans ce chapitre est basée sur les fractions texturales du sol (sables, limons et argiles) et les minéraux que ces fractions contiennent.
Importance de la fraction minérale du sol
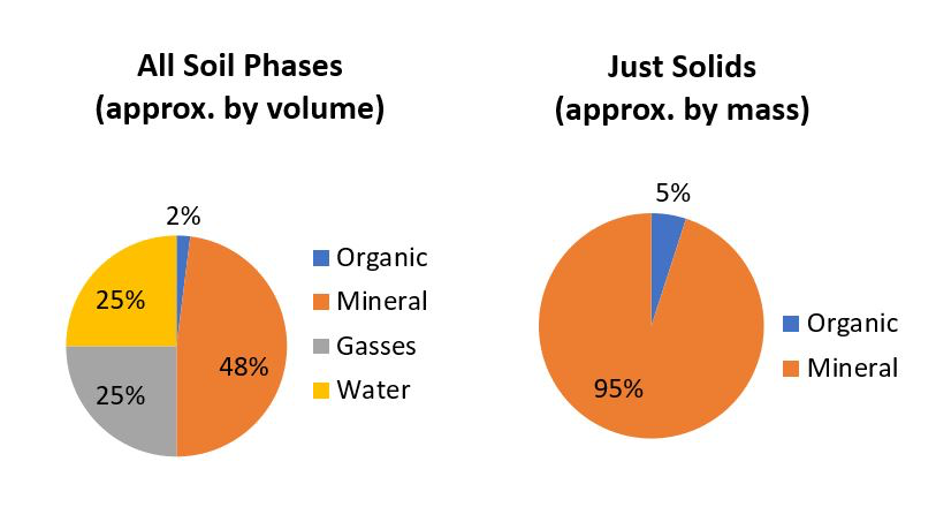
La plupart des sols sont composés de matières minérales qui constituent la majeure partie d’un sol. Les sols bien structurés sont composés d’environ 50 % de solides dont la plupart sont des matières minérales (Figure 14.1), à l’exception des sols organiques (>30% de matière organique).
Il y a environ 4 500 à 5 000 minéraux identifiés sur la Terre (MSA, 1997-2020 ; MSA, 2004-2020 ; Mindat, 2020), la plupart étant soit rares, soit trouvés dans des poches isolées (par exemple, des gisements de minéraux à valeur économique) au plus profond de la croûte terrestre. Les minéraux sont des cristaux inorganiques naturels comprenant une gamme définissable de compositions chimiques. Par exemple, le minéral terrestre le plus répandu dans les sols canadiens est de loin le α-quartz (α-SiO2); prononcé “alpha-quartz”, où alpha fait référence à la forme cristalline. Des fragments de roche se trouvent également dans le sol, mais ils diffèrent des minéraux en ce que les roches sont des « composites de minéraux » constitués de mélanges physiques de plusieurs minéraux. Par exemple, le granite est une roche ignée qui constitue la grande majorité du Bouclier canadien. Le granite est généralement composé d’un mélange de quatre types de minéraux : le α-quartz, les feldspaths, les amphiboles et les micas qui, en raison des actions de broyage et de mélange par les glaciers, sont des minéraux communément trouvés dans nos sols. Les fragments de roches se trouvent parmi les plus grosses particules de sol (c’est-à-dire de la taille de sables ou plus gros). Lors de leur transport, la fragmentation physique des roches en fragments de plus en plus petits se fait le long des bordures des minéraux adjacents. Ces bordures sont les zones les moins stables des roches; ce qui explique que les fragments les plus petits sont représentés par des minéraux individuels distincts.
La plupart des minéraux trouvés dans le sol sont communs, c’est-à-dire très répandus et présents en grandes quantités, et leur valeur économique est généralement très faible. Les types de minéraux trouvés dans le sol ne représentent qu’une petite fraction (<100) de tous les minéraux connus; cependant, lorsqu’ils interagissent avec la solution du sol (l’eau du sol), l’atmosphère et la matière organique du sol, ils ont une influence considérable sur les caractéristiques chimiques et la nature des sols.
Les minéraux du sol sont la source naturelle de nutriments pour les végétaux et sont libérés lentement au cours du temps par l’altération chimique. Toutes les plantes ont besoin d’un minimum de 17 éléments nutritifs pour compléter leur cycle de vie (voir Chapter 7). À l’exception de C, H et O, que les plantes obtiennent de l’air et de l’eau, les plantes dérivent les 14 éléments restants (N, P, K, Ca, S, Mg, Fe, B, Cl, Mn, Zn, Cu, Mo et Ni) principalement à partir des minéraux du sol ou par l’ajout d’engrais, de fumier et d’autres types d’amendement (Parikh et James, 2012 ; Singh et Schulze, 2015). Quatre autres éléments (Na, Si, V et Se) sont essentiels à la croissance de certaines plantes (Havlin et al., 2005). Les minéraux du sol contribuent également à la capacité d’un sol à retenir les éléments nutritifs grâce aux processus d’échanges de cations et d’anions.
MATÉRIAUX PARENTAUX DU SOL
Les sols canadiens sont très jeunes par rapport à ceux des tropiques, qui n’ont jamais été remaniés par les glaciers (voir Chapter 2). Le Canada contient plus de terrains glaciaires que tout autre pays au monde (Rutter, 2015) et presque tous les sols canadiens se sont développés sur des sédiments glaciaires du Wisconsinien supérieur déposés ces derniers 5 000 à 18 000 ans. Les seules exceptions sont certaines petites zones telles que la région de Cypress Hills dans le sud de l’Alberta et de la Saskatchewan, les zones non glaciaires du nord du Yukon ainsi que quelques petites parties de l’ouest des Territoires du Nord-Ouest qui n’étaient pas recouvertes de glace pendant la glaciation du Wisconsinien (voir Chapter 2). Par comparaison, des sols tels que ceux du sud des États-Unis, de l’Australie, d’Afrique et d’Amérique du Sud ont plusieurs millions d’années. Par conséquent, les sols canadiens sont composés de minéraux hérités principalement du substratum rocheux sous-jacent, ainsi que de minéraux déposés par les glaciers lors de l’avancée de la glace, ou lors de la déglaciation par l’eau de fonte et/ou par le vent soufflant sur les grandes plaines dénudées.
MINÉRAUX PRIMAIRES et SECONDAIRES
La croûte lithosphérique peut être décrite dans un contexte très large comme étant composée de deux lithologies principales : la croûte océanique composée majoritairement de roches mafiques et ultramafiques (riches en Fe et Mg) ; et la croûte continentale qui contient davantage de roches felsiques ignées et métamorphiques, et des roches sédimentaires. Puisque les sols sont situés dans des zones continentales, ils se développent principalement à partir des roches de la croûte continentale, qui, dans le contexte canadien, sont constituées d’environ 85% de roches ignées et métamorphiques et de 15% de roches sédimentaires.
Les minéraux dérivés directement des processus géologiques et géochimiques ayant lieu dans la croûte terrestre sont appelés « minéraux primaires ». L’examen des surfaces minérales au microscope peut révéler certains changements dus à l’altération biogéochimique (Spiers et al., 1989), mais la plupart des minéraux primaires des sols canadiens n’ont été que très peu altérés par des actions chimiques au cours des dernières 5 000 à 18 000 années.
Lorsqu’ils sont exposés à la surface de la Terre, la plupart des minéraux primaires sont chimiquement instables. Bien qu’ils persistent dans nos sols, ils finiront, avec le temps (à l’échelle des temps géologiques), par succomber aux processus biogéochimiques de l’environnement et libérer progressivement leurs composants élémentaires dans l’environnement. Les minéraux primaires s’altèrent et se dissolvent à des rythmes différents. En général, les minéraux contenant du Na et du K se dissolvent plus rapidement que ceux contenant du Ca et du Mg, qui à leur tour se dissolvent plus rapidement que les minéraux composés de Si, Al et Fe. Le fer est un cas un peu particulier car c’est le seul élément majeur à deux valences (Fe2+ et Fe3+) et dont la solubilité est très différente selon l’environnement chimique. Dans des conditions réductrices (absence d’oxygène), Fe a tendance à être soluble et présent sous forme d’ion Fe2+. Dans des conditions oxydantes, Fe2+ réagit avec l’oxygène (oxydé) pour former des ions Fe3+ qui à leur tour forment des minéraux oxyhydroxydés insolubles (en fait la composition de la rouille). D’autres éléments mineurs (par exemple Mn, As, U, etc.) sont transformés de la même manière. Le résultat est qu’à mesure que l’altération progresse, les sols s’appauvrissent généralement en Na, K, Ca et Mg qui sont peu à peu lessivés des sols et s’enrichissent en Si, Al et Fe qui eux sont moins lessivés (Figure 14.2). Le lessivage des sols par l’eau explique aussi pourquoi les océans (bassin de réception ultime des eaux continentales) contiennent des concentrations élevées de sels solubles (Na, K, Cl et SO4). Les minéraux carbonatés (principalement CaCO3) se forment ou se dissolvent en fonction du pH de leur solution porteuse. Ils précipitent dans des conditions alcalines et se dissolvent dans des conditions acides. Le CO2 atmosphérique réagit avec l’eau pour produire un acide faible (H2CO3) qui permet la dissolution du carbonate de calcium (et d’autres types de roches). Les eaux souterraines plus profondes ont tendance à être alcalines, favorisant ainsi la précipitation des carbonates généralement trouvés plus profondément dans les profils de sol (horizons C)
.
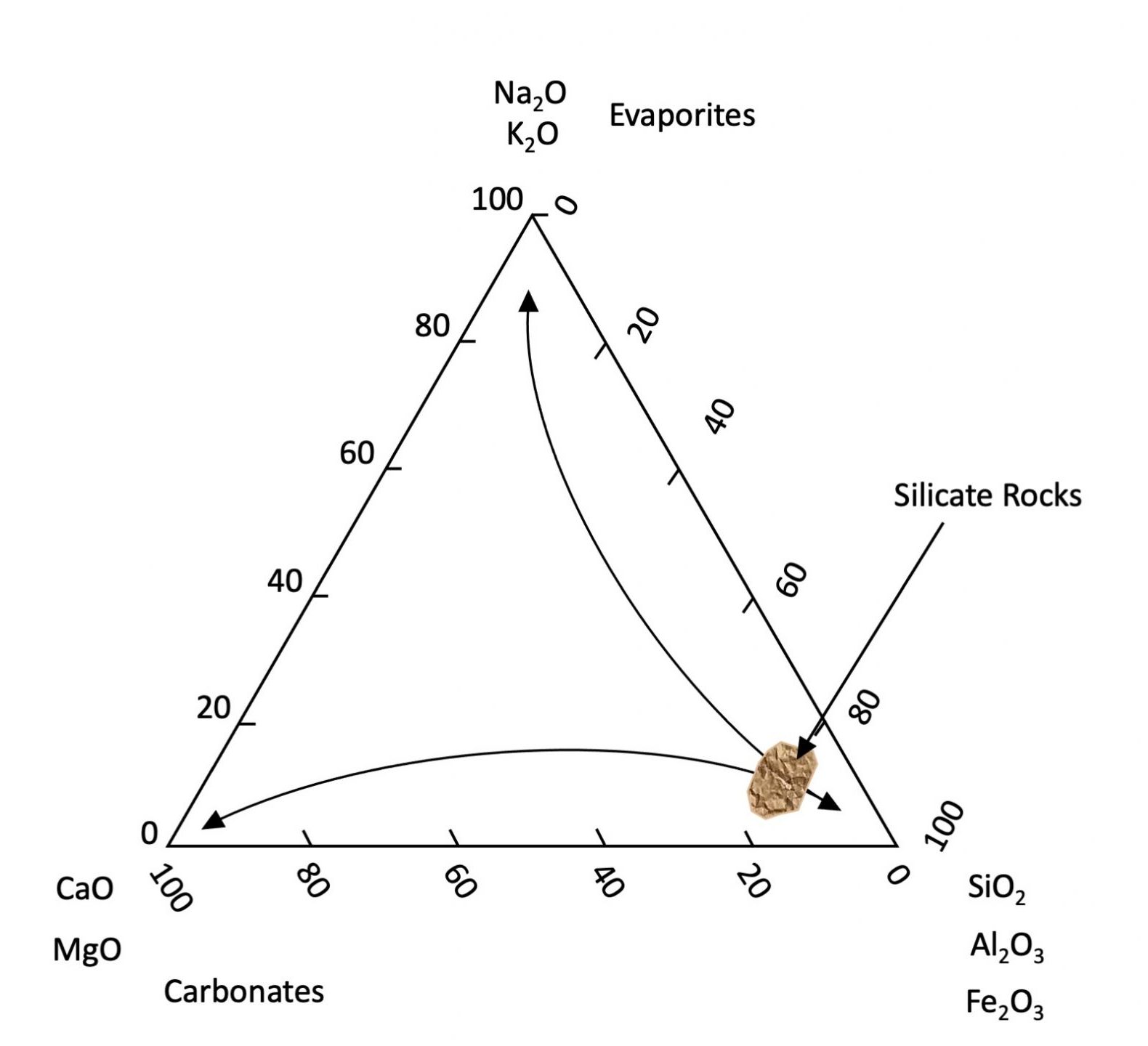
Alors que les minéraux primaires se forment par des processus géologiques, les minéraux secondaires sont un sous-produit de l’érosion chimique. Ils sont issus soit de l’altération chimique partielle des minéraux primaires, soit de la précipitation chimique de composants dissous dérivés d’autres minéraux. La plupart des minéraux secondaires sont des aluminosilicates hydratés ou des oxyhydroxydes de Fe et d’Al qui sont stables dans les sols. La plupart des aluminosilicates hydratés sont classés comme des phyllosilicates ou des minéraux argileux, qui sont les minéraux secondaires les plus abondants dans les sols canadiens. Ce sont des particules en forme de feuillets à grains très fins, caractérisées par un rapport surface/masse, aussi appelé surface spécifique (SS), très élevé. Étant donné que la SS est inversement proportionnelle au diamètre des particules, la fraction argileuse représente de loin la fraction minérale avec la plus grande surface d’échange des minéraux du sol (Chapter 5, Tableau 5.1).
Les minéraux argileux sont également les phases inorganiques les plus réactives chimiquement dans les sols, et la quantité et le type de minéraux argileux influencent de nombreuses propriétés du sol, notamment :
- leur porosité et leur taille des pores;
- leur expansibilité (comportement gonflant);
- leur compressibilité et leur compactibilité;
- leur capacité de sorption;
- leur capacité d’échange; et,
- leur conductivité hydraulique.
Pouvez-vous creuser!
Bien que des minéraux secondaires se forment et soient stables dans les sols à la surface de la Terre, la grande majorité des minéraux que l’on trouve dans les sols canadiens se sont formés dans un passé lointain, avant la fin de la glaciation du wisconsinien. La plupart des minéraux du sol déposé au cours de la dernière glaciation provenaient de sédiments de surface formés et déposés, probablement à plusieurs reprises, au cours des millénaires précédant la dernière glaciation. Bien que des minéraux secondaires continuent de se former dans nos sols en réponse à l’altération chimique, les quantités totales produites depuis le dernier événement glaciaire sont très faibles par rapport aux quantités redéposées lors des nombreuses glaciations du passé
MINÉRAUX et TAILLE DES PARTICULES
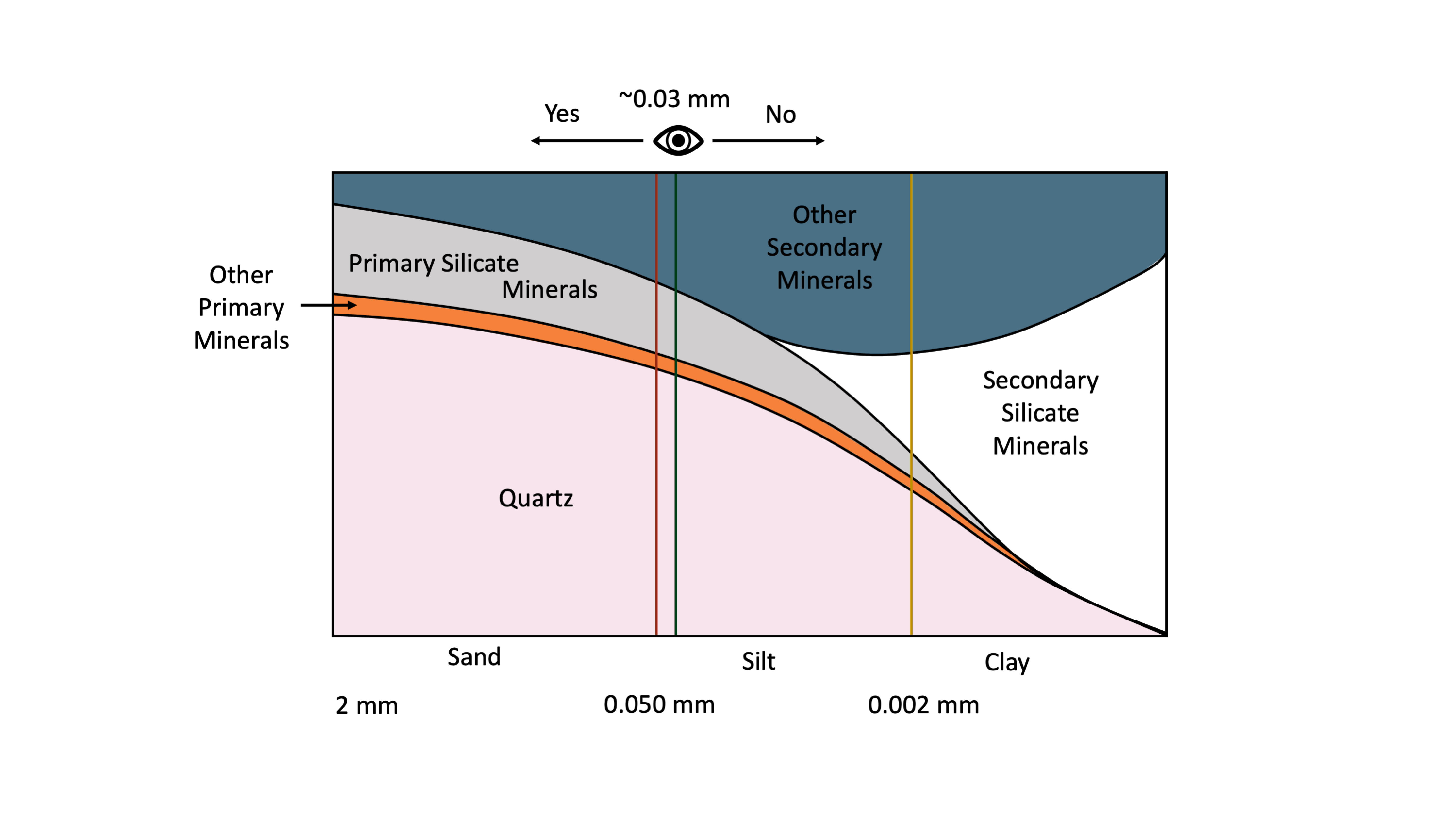
Une personne avec une vision de 20:20 peut distinguer à l’œil nu des particules individuelles aussi petites que ~30 µm (0,03 mm). Il s’agit de la séparation approximative entre les sables et les limons (0,05 mm) (Figure 14.3). Par comparaison, l’épaisseur d’un cheveu humain est comprise entre 0,1 et 0,04 mm. Cela signifie qu’un individu moyen avec une vue raisonnablement bonne peut facilement distinguer à l’œil nu (sans grossissement) les particules individuelles de sable mais pas la plupart des particules individuelles de limons, et certainement pas les particules d’argile. L’observation des particules de limons nécessite un grossissement à l’aide d’un microscope optique, tandis que les particules d’argile ne peuvent être examinées qu’en utilisant la microscopie électronique (Smart et Tovey, 1981) ou la microscopie à force atomique (Liu, 2003).
La distribution des minéraux du sol varie avec la taille des particules (Figure 14.3). Sur la base des fractions granulométriques définies dans le Système Canadien de Classification des Sols (SCWG 1998), la fraction sableuse (particules d’un diamètre compris entre 2 et 0,050 mm) est dominée par les minéraux primaires. Les minéraux primaires sont principalement du quartz, des feldspaths et d’autres minéraux silicatés primaires tandis que la fraction argileuse (<0,002 mm de diamètre) est dominée par des phyllosilicates secondaires et des oxyhydroxydes de fer et d’aluminium. La fraction limoneuse (0,05-0,002 mm de diamètre) est de taille intermédiaire entre les sables et les argiles et contient généralement un mélange de minéraux primaires et secondaires.
Abondance élémentaire et structures minérales
Il existe 91 éléments naturels, qui se trouvent tous à certaines concentrations dans le sol. Le tableau 14.1 fournit une liste de 70 éléments par ordre d’abondance décroissante dans les roches crustales et dans les sols. La plupart des éléments se substituent les uns aux autres dans la structure cristalline des minéraux en fonction de leur taille et de leur valence. Ceci sera discuté en détail dans ce chapitre. En fonction de la solubilité et de la résistance à l’altération chimique, la teneur de certains éléments contenus dans les minéraux du sol augmente par rapport à l’abondance dans la croûte continentale (par exemple Si et O), tandis que la concentration de la plupart des éléments diminue avec le temps (comparez l’abondance élémentaire de la croûte avec l’abondance du sol ; Tableau 14.1).
Table 14.1. Teneur totale (mg kg-1) en éléments dans les roches de la croûte terrestre continentale et dans les sols (Adapté de Bowen, 1979). Les éléments sont classés par ordre d’abondance décroissante
| Élément | Croûte | Sol | Élément | Croûte | Sol | |
|---|---|---|---|---|---|---|
| O | 464000 | 490000 | B | 10 | 38 | |
| Si | 281500 | 330000 | Th | 9.6 | 14 | |
| Al | 81650 | 71000 | Sm | 6 | 6 | |
| Fe | 53000 | 32000 | Gd | 5.4 | 3.5 | |
| Ca | 38650 | 15000 | Cs | 3 | 3 | |
| Na | 26150 | 10900 | Dy | 3 | 5.7 | |
| K | 23950 | 18300 | Yb | 3 | 3.9 | |
| Mg | 21950 | 8300 | Hf | 3 | 7.7 | |
| Ti | 5050 | 5100 | Be | 2.8 | 1.5 | |
| P | 1050 | 800 | Er | 2.8 | 3 | |
| Mn | 950 | 760 | U | 2.7 | 2.2 | |
| F | 625 | 270 | Br | 2.5 | 43 | |
| Ba | 425 | 568 | Sn | 2 | 5.8 | |
| Sr | 375 | 278 | Ta | 2 | 1.2 | |
| S | 260 | 433 | As | 1.8 | 11 | |
| Zr | 165 | 345 | Ge | 1.5 | 3 | |
| V | 135 | 108 | Mo | 1.5 | 1.9 | |
| Cl | 130 | 485 | W | 1.5 | 1.1 | |
| Cr | 100 | 84 | Eu | 1.2 | 1.3 | |
| Rb | 90 | 120 | Ho | 1.2 | 0.8 | |
| Ni | 75 | 34 | Tb | 0.9 | 0.85 | |
| Zn | 70 | 60 | I | 0.5 | 7.1 | |
| Ce | 60 | 84 | Lu | 0.5 | 0.46 | |
| Cu | 55 | 26 | Tm | 0.48 | 0.62 | |
| Y | 33 | 28 | Tl | 0.45 | 0.25 | |
| La | 30 | 41 | Cd | 0.2 | 0.6 | |
| Nd | 28 | 44 | Sb | 0.2 | 1.7 | |
| Co | 25 | 12 | Bi | 0.17 | 0.5 | |
| Sc | 22 | 10 | In | 0.1 | 1 | |
| Li | 20 | 31 | Hg | 0.08 | 0.1 | |
| Nb | 20 | 14 | Ag | 0.07 | 0.05 | |
| Ga | 15 | 21 | Se | 0.05 | 0.4 | |
| Pb | 12 | 29 | Pt | 0.001 | 0.001 | |
| Pr | 11 | 6.5 | Au | 0.001 | 0.001 | |
| Remarque: Le tableau n’inclut pas l’hydrogène, le carbone, l’azote, les gaz nobles ou les éléments trouvés en quantités d’ultra-traces (<0.001 mg kg-1). Voir https://earthref.org/GERMRD/10/ pour la version la plus récente de ce tableau. Voir Spiers et al. (1989a) pour les éléments des sols de l’Alberta. | ||||||
Un examen plus approfondi des abondances élémentaires indique que les huit éléments les plus abondants (O, Si, Al, Fe, Ca, Na, K et Mg) représentent la quasi-totalité de la masse (99 %) de la croûte terrestre (Tableau 14.2). En outre, lorsqu’on considère les rayons atomiques, on voit que près de 85% de la croûte terrestre en volume est composée d’oxygène sous la forme d’oxydes. Plus précisément, les minéraux à base d’oxyde (O2-) forment la structure fondamentale de la plupart des minéraux de la croûte terrestre, les principaux cations (Si, Al, Fe, Ca, Na, K et Mg) s’insérant entre les atomes d’oxygène au niveau atomique.
Table 14.2. Rayons du O2- et des cations majeurs, abondance dans la croûte, ratios des rayons et nombres de coordinations prédits
| Élément | Rayon atomique (nm) | Abondance massique (%) | bondance volumique (%) | Ratio des rayons | Nombre de coordinations prédit (NC) |
|---|---|---|---|---|---|
| O2- | 0.14 | 0.464 | 0.845 | na* | na* |
| Si4+ | 0.04 | 0.282 | 0.012 | 0.29 | 4 |
| Al3+ | 0.054 | 0.0817 | 0.0083 | 0.38 | 4 |
| Fe3+ | 0.065 | 0.053 | 0.0094 | 0.46 | 6 |
| Fe2+ | 0.078 | Incl.** | Incl.** | 0.56 | 6 |
| Ca2+ | 0.112 | 0.0387 | 0.0361 | 0.80 | 8 |
| Na+ | 0.097 | 0.0262 | 0.0285 | 0.69 | 6 |
| K+ | 0.151 | 0.022 | 0.0548 | 1.08 | 12 |
| Mg2+ | 0.072 | 0.024 | 0.0054 | 0.51 | 6 |
| Total | 0.9909 | 1 | |||
| Remarque: *na = non applicable. **: l’abondance du Fe2+ est incluse avec Fe3+. | |||||
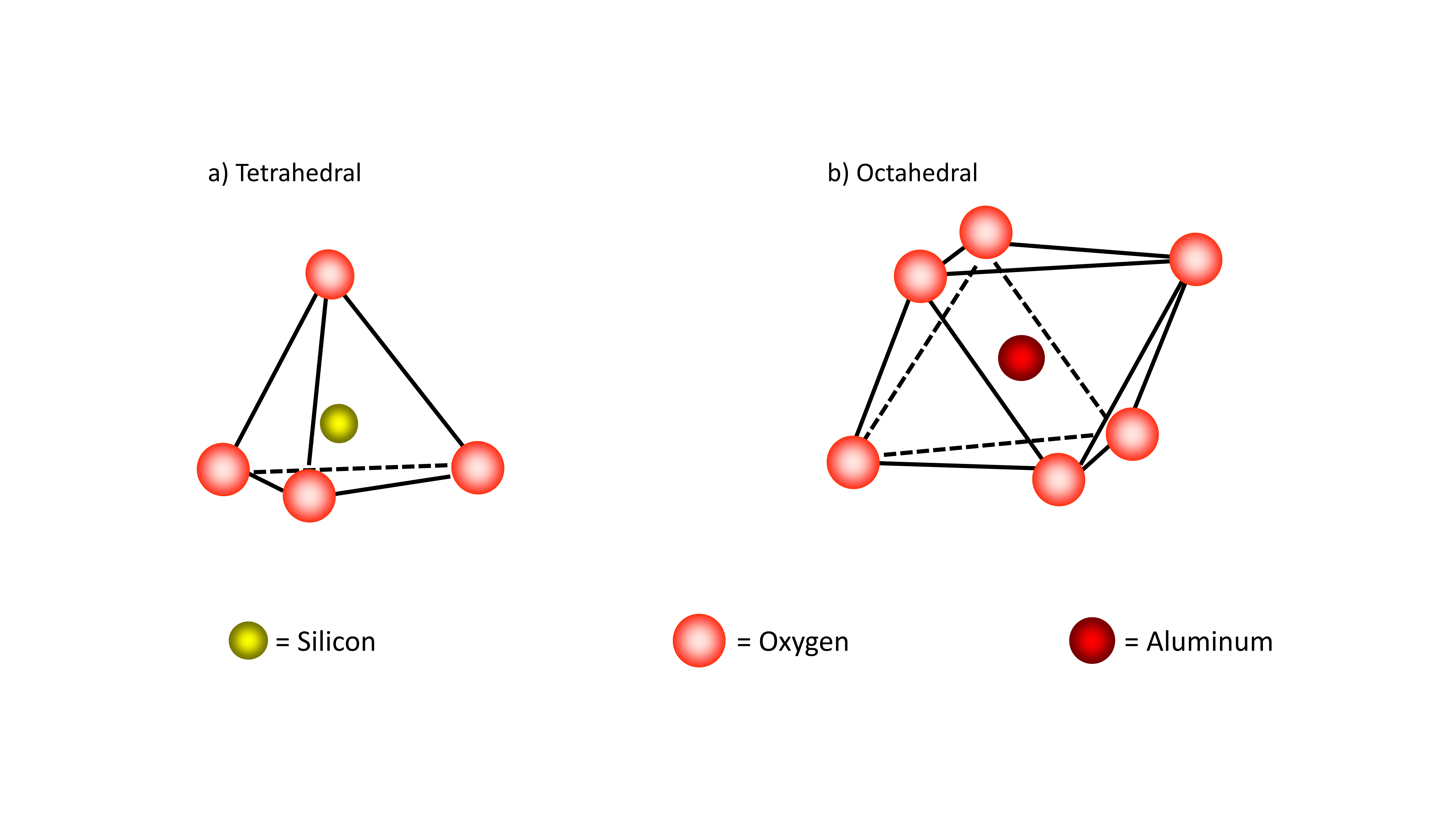
La structure cristalline des minéraux au niveau atomique peut être facilement visualisée si l’on considère que l’oxygène et les autres ions métalliques agissent comme des sphères rigides avec des rayons fixes. Bien que cela ne soit pas strictement vrai, il s’agit d’un concept utile pour visualiser les structures cristallines. La géométrie de ces sphères rigides est régie par les règles de Pauling (Pauling 1929) dont la première stipule que la distance entre un cation et un anion est la somme de leurs rayons. Les ions dans une structure cristalline ont tendance à rassembler autour d’eux autant d’ions de charges opposées que leur taille le permet. Le nombre d’ions O2- qui peuvent entourer un cation central est appelé le nombre de coordination (NC). Les gros cations (par exemple K+) ont des nombres de coordination élevés en raison de leurs grands volumes, tandis que les petits cations (par exemple, Si4+) ont des nombres de coordination faibles (Figure 14.4). Notez également que les cations combinant un grand rayon atomique et une faible valence (par exemple K+ et Na+) sont les plus mobiles tandis que ceux avec des rayons plus petits et une charge élevée (Si4+, Al3+ et Fe3+) sont beaucoup moins mobiles.

Étant donné que les ions ont tendance à s’entourer d’autant d’ions de charges opposées que possible, on peut prédire les nombres de coordination uniquement à partir des tailles relatives des ions, exprimées sous forme de ratio des rayons :
(1) 
Le tableau 14.3 fournit les gammes de ratios de rayons, les nombres de coordination et les unités structurelles correspondants. Certains chevauchements et incertitudes surviennent parce que l’hypothèse que les ions sont comme des sphères rigides avec des rayons constants n’est pas strictement vraie. Dans cet esprit, les ions sont généralement interchangeables si leur taille ne diffère pas d’environ 15 % et plus, et que leur valence ne diffère pas de plus d’une unité. En réalité, les ions ne sont pas sphériques, et leurs rayons et leurs nombres de coordination changent légèrement. Par conséquence, certains éléments ont communément plus d’une géométrie de coordination. Par exemple, l’aluminium se trouve couramment dans des unités tétraédriques (NC=4) et octaédriques (NC=6). La substitution de cations dans les structures minérales est appelée substitution isomorphe et sera discutée plus en détail plus loin dans ce chapitre.
La géométrie des ions autour d’un ion central peut être représentée sous la forme d’un polyèdre de coordination. En minéralogie, ces polyèdres sont généralement construits à partir de l’arrangement d’anions autour de cations, mais les cations peuvent parfois être totalement absents. Le tétraèdre (structure à quatre côtés avec quatre atomes d’oxygène se coordonnant autour de Si) et l’octaèdre (structure à huit côtés avec six atomes d’oxygène se coordonnant autour de l’aluminium; Figure 14.5) sont de loin les structures polyédriques composant la plupart des minéraux du sol les plus courants. Étant donné que Si et O sont de loin les éléments les plus abondants dans le sol, les minéraux à structure tétraédrique constituent la base de nombreux minéraux primaires communs. Les phyllosilicates feuilletés composés d’une combinaison de tétraèdres de silice et d’octaèdres d’aluminium sont les minéraux secondaires dominant la fraction argileuse (<0,002 mm). Les phyllosilicates seront discutés plus en détail plus loin dans ce chapitre.
Table 14.3. Ratios des rayons, nombres de coordination et unités de structure minérale vis-à-vis de l’oxygène structural (O2-) correspondantes pour la plupart des cations
| Gamme de Rcation/R(O2-) | Nombre de Coordination (NC)) | Unité polyédrique |
|---|---|---|
| 0.155-0.225 | 3 | Triangulaire |
| 0.225-0.414 | 4 | Tétraédrique |
| 0.414-0.732 | 6 | Octaédrique |
| 0.732-1 | 8 | Cubique |
| >1 | 12 | Cuboctaèdre |
La différence fondamentale entre les structures minérales est le nombre d’atomes d’oxygène partagés entre les tétraèdres adjacents. Bien qu’il existe plusieurs types de structures et de classifications minérales, les minéraux du sol les plus courants sont de manière générale composés d’unités répétitives de tétraèdre de silicate (SiO4) qui entrent dans l’un des quatre types généraux de structures simplifiées :
- des tétraèdres de silice isolés (néosilicates) ;
- chaînes de tétraèdres de silice (inosilicates) ;
- structure de tétraèdres (tectosilicates) ; et,
- feuillets de tétraèdres (associées à des unités octaédriques; phyllosilicates).
Certains minéraux communs du sol sont indiqués dans le tableau 14.4 et répertoriés en fonction de la quantité d’atomes d’oxygène partagés entre les tétraèdres adjacents.
Table 14.4. Quelques-uns des minéraux les plus communs du sol
| Partage d’oxygène dans les tétraèdres | Groupes minéraux communs | Exemple de minéraux | Composition idéale généralisée |
|---|---|---|---|
| Néosilicates | Olivines | Fostérite | Mg2SiO4 |
| Pas de partage d’oxygène | |||
| Tétraèdres isolés | Fayalite | Fe(II)2SiO4 | |
| Inosilicates | Pyroxènes | Enstatite | MgSiO3 |
| Un oxygène en partage | Diopside | (Ca,Mg)SiO3 | |
| Augite | (Ca,Mg,Fe(II),Al)SiO3 | ||
| Inosilicates | Amphiboles | Trémolite | Ca2Mg5[Si8O22(OH)2] |
| Deux oxygènes en partage | Actinolite | Ca2(Mg,Fe(II))5[Si8O22(OH)2] | |
| Hornblende | (Ca,Na)(Mg,Fe(II),Al)5[(Al,Si)8O22(OH)2] | ||
| Phyllosilicates | Kaolinite | Si2Al2O5(OH)4 | |
| Trois oxygènes en partage | Muscovite | KAl3(AlSi3)O10(OH)2 | |
| Tectosilicates : les quatre oxygènes en partage | Structure de tétraèdres | Quartz | α-SiO2 |
| Feldspaths | (Ca,Na,K)(AlxSi(3-x))O8 |
MINÉRAUX DE LA FRACTION SABLEUSE (2 mm – 0.05 mm)
Quartz
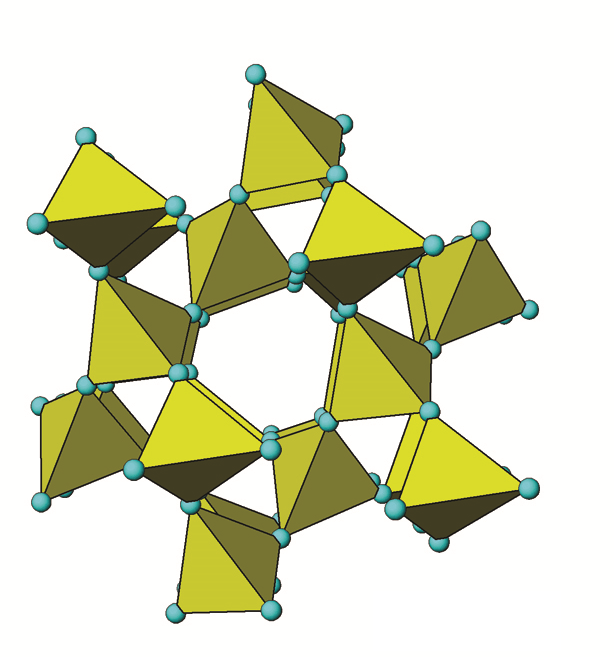
Le quartz (SiO2), ou plus précisément le α-quartz (α-SiO2), est le minéral le plus courant dans les sols canadiens, représentant généralement la majorité de la fraction sableuse (Figure 14.3). Le quartz est un tectosilicate (un silicate de structure) dont les quatre atomes d’oxygène des tétraèdres de silice sont partagés avec les tétraèdres Al, Si3 adjacents (Figure 14.6). Il appartient au système cristallin hexagonal.
Lorsque les tétraèdres partagent tous les atomes d’oxygène avec les tétraèdres adjacents, ils forment une structure tridimensionnelle très solide de liaisons Si-O. Le quartz est essentiellement du SiO2 pur. La charge négative des atomes de O est exactement équilibrée par les atomes de Si, ne nécessitant aucun autre ion de liaison. Au microscope, la plupart des spécimens sont généralement des fragments brillants de forme irrégulière à l’aspect blanc, laiteux et parfois clair. La structure chimique en 3D du quartz est typique des tectosilicates. SiO2 est l’unité de base, qui est répliquée dans une configuration tétraédrique. Les liaisons Si-O sont des liaisons covalentes très fortes et très résistantes à l’altération. La solubilité du quartz est donc très faible et son taux d’altération (dissolution) extrêmement lent. Ceci le rend très persistant dans le sol par rapport à d’autres minéraux, ce qui conduit à son accumulation relative au fil du temps (par exemple, dans les horizons Ae des sols podzoliques et luvisoliques). À l’œil, le quartz apparaît généralement sous forme de fragments vitreux clairs, blancs ou gris, mais il existe parfois sous forme de fragments jaunes, bruns, violets, roses ou rouges qui peuvent facilement être vus à l’œil nu ou à la loupe. Le quartz constitue généralement environ 40 à 60% de la fraction sableuse (2,0 à 0,05 mm), une grosse proportion de la fraction limoneuse (0,05 à 0,002 mm) et une faible proportion de la fraction argileuse (<0,002 mm) des sols canadiens (Figure 14.3).
Feldspaths
Les feldspaths sont après le quartz le deuxième groupe de tectosilicate le plus répandu dans le sol, représentant généralement 15 à 35 % de la fraction sableuse (Figure 14.3). Les feldspaths constituent une grande partie des roches granitiques et métamorphiques. Par conséquent, ils constituent une partie importante de notre sol. Il existe de nombreuses espèces minérales composant le groupe des feldspaths, toutes ayant une formule générale de la forme X(Al,Si)4O8, où X est le plus souvent K, Na ou Ca, et les ions Al et Si constituent les unités tétraédriques. D’autres ions tels que Ba, Zn, Rb, Sr et Fe remplacent également X dans les structures de feldspath (King 2020). Les feldspaths apparaissent généralement blanc cassé, avec diverses nuances de rouge, d’orange, de brun et parfois de vert. La structure tridimensionnelle des feldspaths est constituée d’unités allant de AlSi3O8 à Al2Si2O8 dans une configuration en « ruban coudés » (Figure 14.7a). La charge négative de la structure, causée par la substitution de certains atomes de Si par Al dans la structure des coudes, est neutralisée par des cations K, Na ou Ca qui se logent dans les vides des coudes.
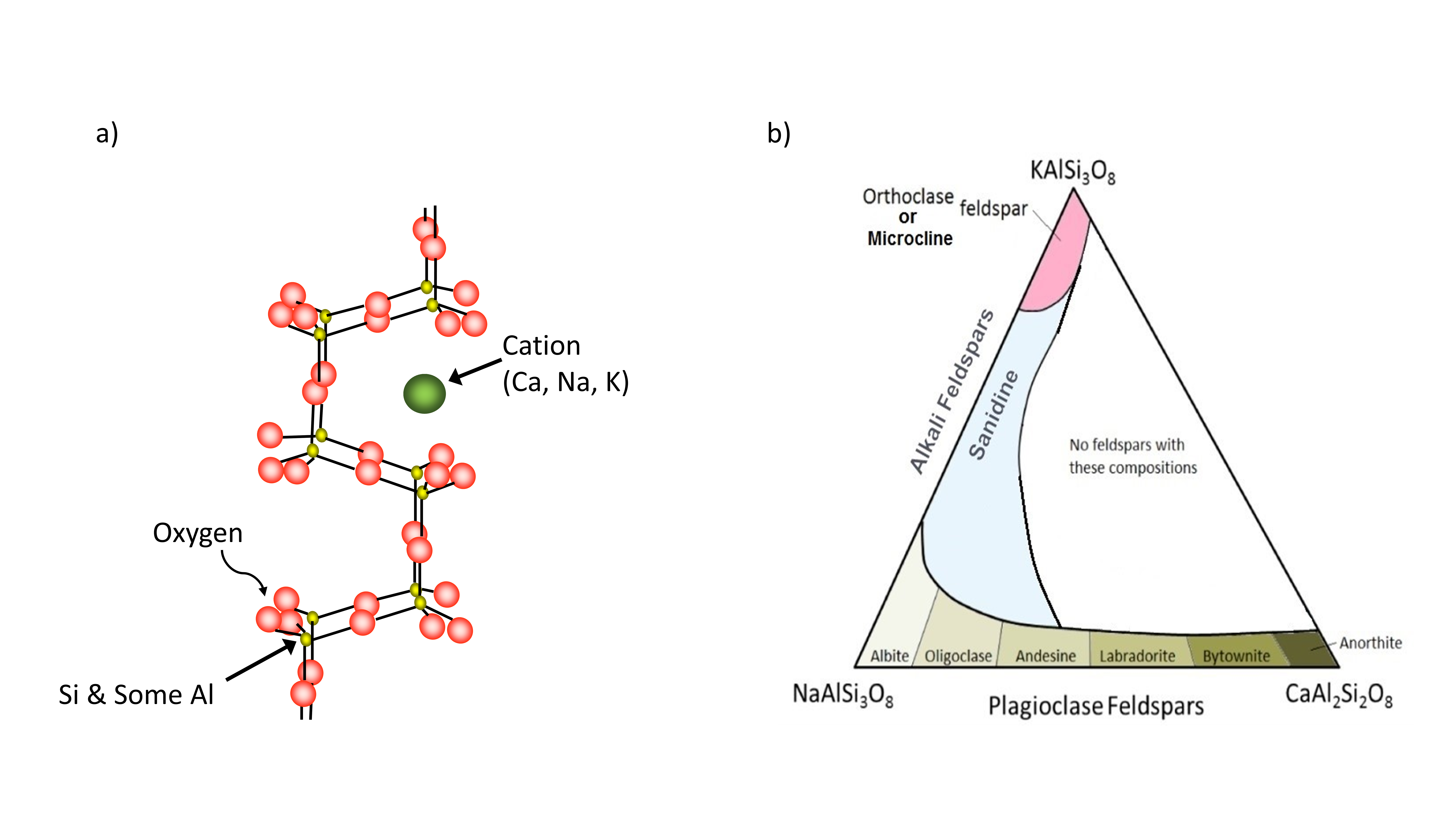
Les minéraux de feldspath les plus courants et leurs gammes de composition sont illustrés dans le diagramme ternaire de la Figure 14.7b. Orthoclase et microcline sont les noms de minéraux donnés aux feldspaths ayant une composition chimique similaire et dont la structure est dominée par le potassium (KAlSi3O8) accompagné de quantités mineures (jusqu’à environ 25%) de Na à la place de K (Figure 14.7B). L’orthose et la microcline sont polymorphes, ce qui signifie qu’ils ont la même composition chimique mais des formes cristallines différentes. L’orthoclase a une forme cristalline monoclinique tandis que le microcline est triclinique. Les feldspaths contenant plus de 25 % de Na mais moins de 75 % de K constituent le minéral sanidine [(K,Na)AlSi3O8]. Ces feldspaths, quelle que soit leur composition en K et Na (orthoclase/microcline, sanidine à albite) sont généralement appelés feldspaths alcalins (Figure 14.7B). Les feldspaths sodiques et calciques appartiennent à une série continue appelée la série des plagioclases. Leur composition va de NaAlSi3O8 à CaAl2Si2O8, et la variation de charge résultant de la quantité de Na et de Ca dans la structure est neutralisée par substitution d’Al par Si dans les coudes tétraédriques. Les feldspaths plagioclases sont nommés spécifiquement à leur composition (tableau 14.5). La série de minéraux des plagioclases est un exemple de série de solution solide dans laquelle la composition chimique varie entre deux pôles compositionnels (deux minéraux situés aux extrémités de la série; c’est-à-dire, albite; NaAlSi3O8 et anorthite; CaAl2Si2O8) qui partagent la même formule chimique de base mais ont avec des substitutions élémentaires différentes à un ou plusieurs sites atomiques.
Le quartz et les feldspaths ont une densité et une dureté similaires, ils ont donc tendance à être transportés et déposés ensemble. Contrairement au quartz, les feldspaths ont plusieurs plans de clivage le long desquels les minéraux se fracturent, ce qui les rend cassants et plus sujets à la rupture mécanique lors d’impacts au moment du transport physique. Ces plans de clivage sont des plans de faiblesse le long desquels l’eau et les exsudats microbiens peuvent s’infiltrer et accélérer leur altération chimique. Des organismes tels que des bactéries et des hyphes fongiques infiltrent également ces plans de faiblesse et accélèrent la dégradation des feldspaths. Par conséquent, les feldspaths ont tendance à s’altérer et à se dissoudre plus rapidement que le quartz (Cousineau, 2020).
Table 14.5. Gamme de composition des feldspaths plagioclases
| Nom du minéral | % NaAlSi3O8 | % CaAl2Si2O8 | Gamme de Composition |
|---|---|---|---|
| Albite | 90-100 | 0-10 | Na(Al,Si3O8) – Na0.9Ca0.1(Al1.1Si2.9O8) |
| Oligoclase | 70-90 | 10-30 | Na0.9Ca0.1(Al1.1Si2.9O8) – Na0.7Ca0.3(Al1.3Si2.7O8) |
| Andésine | 50-70 | 30-50 | Na0.7Ca0.3(Al1.3Si2.7O8) - Na0.5Ca0.5(Al1.5Si2.5O8) |
| Labradorite | 30-50 | 50-70 | Na0.5Ca0.5(Al1.5Si2.5O8) - Na0.3Ca0.7(Al1.7Si2.3O8) |
| Bytownite | 10-30 | 70-90 | Na0.3Ca0.7(Al1.7Si2.3O8) - Na0.1Ca0.9(Al1.9Si2.9O8) |
| Anorthite | 0-10 | 90-100 | Na0.1Ca0.9(Al1.9Si2.9O8) - Ca(Al2Si2O8) |
Minéraux “lourds”
Les minéraux « lourds » représentent un groupe généralisé d’« autres » minéraux primaires que l’on ne trouve habituellement qu’en petites quantités (2 à 5 %) dans la fraction sableuse des sols canadiens. Plusieurs de ces minéraux sont de couleur foncée en raison de leur teneur élevée en Fe et Mg. D’autres comme le grenat et l’hématite sont rouge foncé, et le zircon est jaune à incolore. Les minéraux lourds peuvent être isolés des fractions sableuses et limoneuses par le biais d’une séparation par densité (Andò, 2020). Les minéraux lourds ne se trouvent généralement pas dans la fraction argileuse. La plupart des minéraux lourds sont généralement définis comme ayant une densité de particules (gravité spécifique) supérieure à 2,90 g/cm3 (Andò, 2020). Certains minéraux communément trouvés dans la fraction sableuse des sols sont listés dans le tableau 14.6. Bien que les minéraux lourds ne soient qu’un composant minéral mineur de la fraction sableuse, leur altération progressive peut fournir de nombreux éléments nutritifs essentiels aux plantes.
Une fois isolée, la fraction minérale lourde peut être examinée par le biais de plusieurs méthodes, notamment la microscopie optique et électronique couplée à des techniques d’apprentissage automatique par ordinateur (Maitre et al., 2019), mais aussi par diffraction des rayons X sur poudre et d’autres méthodes physiques ou chimiques. Les minéraux lourds comprennent généralement des minéraux mafiques avec des concentrations typiquement élevées en Fe et Mg. Macroscopiquement, les pyroxènes et les amphiboles peuvent être difficiles à distinguer les uns des autres, car ils sont tous deux de couleur sombre. En règle générale, les cristaux de pyroxène sont des cristaux plus courts, tandis que les amphiboles forment des cristaux plus longs en forme d’aiguilles (aciculaires). D’autres minéraux dans la fraction minérale lourde peuvent inclure des grenats (X3Al2Si3O12 où X peut être Mg, Fe(II), Mn ou Ca), la magnétite (Fe3O4), la chromite (Fe,Cr(III)2O4), l’hématite (Fe2O3), le zircon (ZrSiO4) et le rutile (TiO2).
Table 14.6. Exemples de minéraux communs de la fraction sableuse avec leurs densités
| Gamme de densité | Minéraux | Densité (g cm-3) |
|---|---|---|
| Fraction “légère” (Densité <2.9 g cm-3) |
Quartz | 2.65 |
| Feldspaths | 2.55-2.76 | |
| Fraction “lourde” (Densité >2.9 g cm-3) |
Olivine | 3.22-4.39 |
| Augite (pyroxène) | 2.96-3.52 | |
| Hornblende (amphibole) | 3.02-3.45 | |
| Hématite (Fe2O3) | 5.26 | |
| Magnétite (Fe3O4) | 5.2 |
MINÉRAUX DE LA FRACTION LIMONEUSE (0.05 – 0.002 mm)
La fraction limoneuse des sols est intermédiaire en taille et en minéralogie aux fractions sableuse et argileuse. La fraction limoneuse contient donc un mélange physique de minéraux trouvés à la fois dans la fraction sableuse et argileuse. Les lecteurs sont donc invités à lire les sections de ce chapitre qui se rapportent aux minéraux de cette fraction pour une discussion plus approfondie.
Pouvez-vous Creuser!
Les termes « argiles » et « minéraux argileux » sont souvent utilisés de manière interchangeable et sont répandus dans la littérature scientifique ; cependant, leur utilisation peut être ambiguë et parfois trompeuse. Le terme « argile » est généralement utilisé pour désigner un sol argileux ou sa fraction argileuse (particules <0,002 mm de diamètre). Les « minéraux argileux » se réfèrent spécifiquement aux phyllosilicates qui représentent la majorité de « l’argile » du sol mais n’incluent pas les oxyhydroxydes et les aluminosilicates amorphes. Bien qu’il ne s’agisse pas de phyllosilicates, de nombreux minéraux de la fraction sableuse décrits ci-dessus peuvent également être présents dans la fraction argileuse.
MINÉRAUX DE LA FRACTION ARGILEUSE (<0.002 mm)
Comme nous l’avons mentionné précédemment, la fraction argileuse est composée principalement d’aluminosilicates hydratés secondaires. Ces minéraux sont à grains très fins et ont donc une SS (surface spécifique) très élevée, et représentent la plus grande partie de la surface minérale disponible et réactive dans le sol (Chapter 5, Tableau 5.1). De plus, les minéraux de la fraction argileuse ont une charge permanente qui varie avec le pH, ce qui les rend capable d’attirer et de retenir les éléments nutritifs pour les plantes avec des impacts directs et indirects sur la fertilité du sol. En bref, ce sont les phases inorganiques les plus chimiquement actives du sol.
SILICATES STRATIFIÉS: PHYLLOSILICATES
Les silicates stratifiés ou en feuillets, appelés phyllosilicates (du grec ancien phyllon signifiant feuille), sont de loin le type de minéral le plus courant dans la fraction argileuse. Ils apparaissent sous forme de structures en feuillets plates dans les microphotographies (par exemple, Beutelspacher et Van Der Marel, 1968 ). Les phyllosilicates sont des minéraux cristallins, avec une structure constituée de couches d’unités atomiques répétitives; d’où leur nom d’aluminosilicates « en couches » ou « feuillets ». Les couches fondamentales sont composées de feuillets tétraédriques et octaédriques qui se présentent sous différentes combinaisons pour former différentes espèces minérales de phyllosilicates.
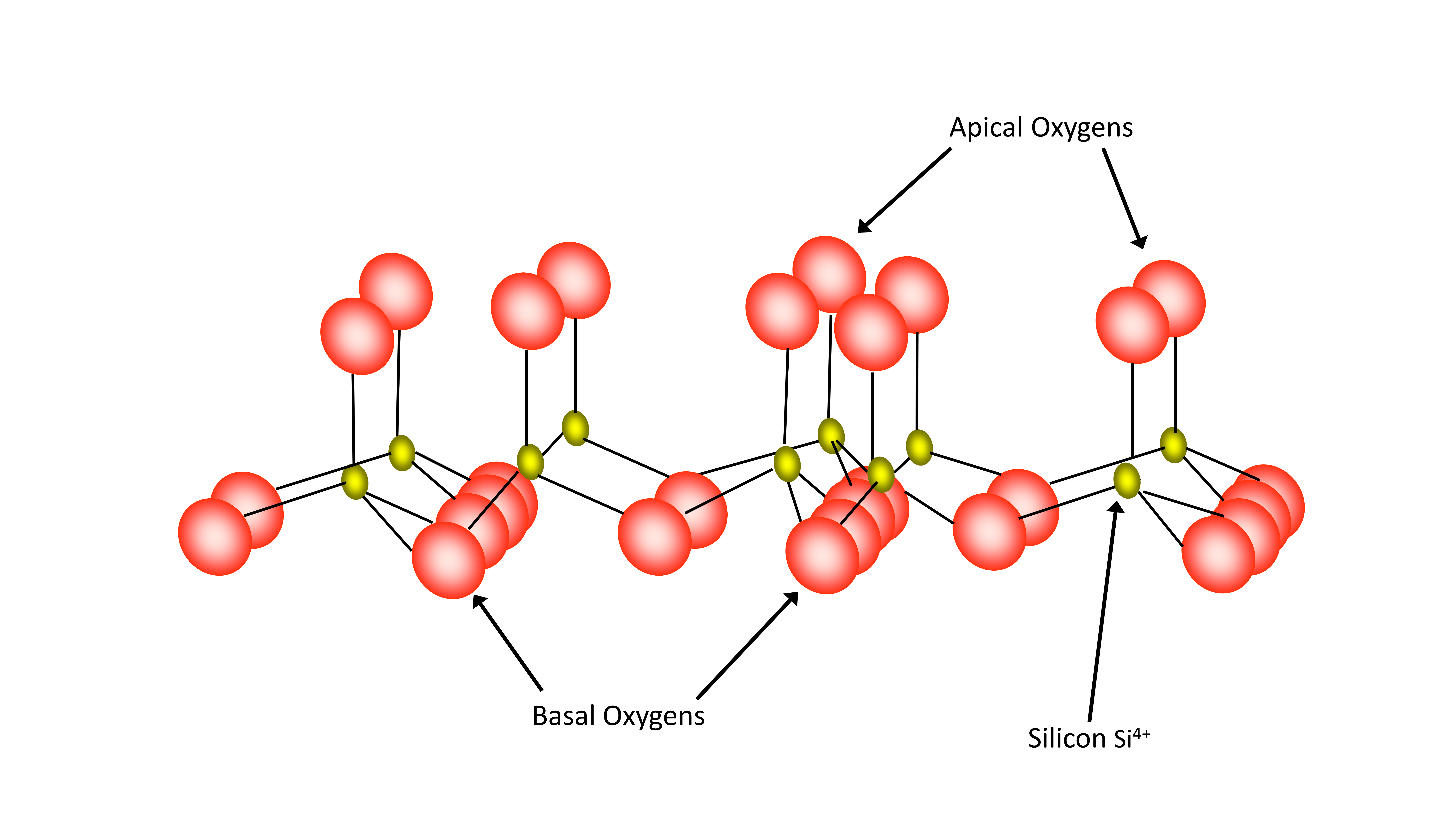
Les feuillets tétraédriques sont composés d’unités répétitives de tétraèdre (à quatre côtés) contenant chacune un atome de Si entouré de quatre atomes d’O. Trois des quatre atomes d’O sont partagés avec des unités tétraédriques adjacentes, appelées atomes O basaux. L’atome d’O qui n’est pas partagé est appelé O apical (Figure 14.8). Les atomes d’O basaux partagés se combinent pour former des feuillets qui se poursuivent en deux dimensions ad infinitum.
Les feuillets octaédriques trouvés dans les phyllosilicates sont composés de plusieurs unités d’octaèdre, chaque unité contenant des atomes d’Al et/ou de Mg entourés de six atomes d’O ou groupes OH-. Trois atomes d’O (ou groupes OH-) se trouvent dans le plan inférieur, et trois atomes d’O (ou groupes OH-) dans le plan supérieur, prenant en sandwich des atomes d’aluminium ou de magnésium formant une configuration à huit côtés (octaédrique) et une forme pseudohexagonale. Ces unités se combinent en partageant des atomes d’O pour former un feuillet octaédrique s’étendant en deux dimensions à l’infini (Figure 14.9). Lorsque le cation central de l’octaèdre est trivalent, comme l’aluminium (Al3+), seuls deux des trois sites cationiques (2/3) d’un feuillet octaédrique doivent être remplis afin de neutraliser la charge des atomes d’O ou des ions OH– environnants. Dans ce cas, le feuillet octaédrique est appelé dioctaèdre (Figure 14.9). Lorsque le cation central de l’octaèdre est divalent, tel que le Mg2+, les trois sites de cations (3/3) d’un feuillet octaédrique doivent être remplis afin de neutraliser la charge négative de l’O ou de l’OH- environnant. Dans ce cas, le feuillet octaédrique est appelé trioctaèdre (Figure 14.9).
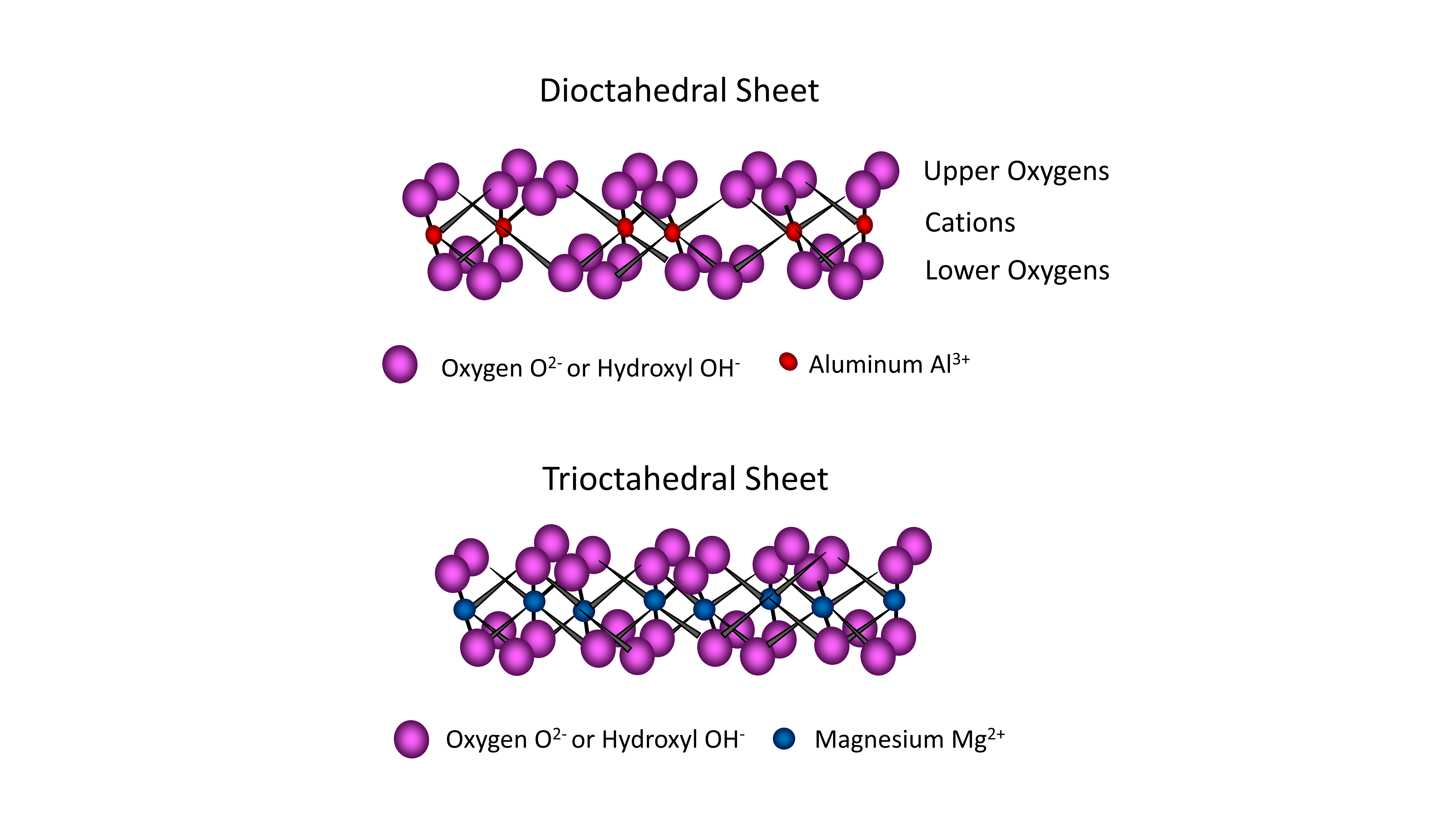
Les structures des phyllosilicates consistent en une combinaison de feuillets tétraédriques et octaédriques par le partage de l’atome O apical du feuillet tétraédrique avec des atomes O du feuillet octaédrique. Les phyllosilicates composés d’un feuillet tétraédrique et d’un feuillet octaédrique sont appelés phyllosilicates de type 1:1. Ceux composés de deux feuillets tétraédriques prenant en sandwich un feuillet octaédrique sont appelés phyllosilicates de type 2:1. Les phyllosilicates se composent de plusieurs couches (de type 1:1 ou de type 2:1) empilées les unes sur les autres et stabilisées par une sorte de liaison chimique. La région située entre deux couches 1:1 ou 2:1 adjacentes est appelée « région intercalaire ». Certains phyllosilicates de type 2:1 peuvent également contenir un feuillet octaédrique supplémentaire composé entièrement d’hydroxydes dans la région intercalaire. Ceux-ci sont appelés phyllosilicates de type 2:1:1 ou 2:2. Le comportement de base et les propriétés des différents phyllosilicates sont dictés en grande partie par les caractéristiques de la région intercalaire des argiles d’un sol donné.
Substitution Isomorphe
En plus de leur SS extrêmement élevée, les phyllosilicates sont également chargés électriquement en raison de la substitution des cations centraux des feuillets tétraédriques ou octaédriques par d’autres cations de taille similaire mais de valence différente (Tableau 14.2). Le tableau 14.3 fournit des gammes de ratios de rayons correspondant aux nombres de coordination communs et à leurs unités structurelles correspondantes. Les ions sont généralement interchangeables au sein d’une structure si leur taille ne diffère pas de plus d’environ 15 % et leur valence de plus d’une unité. La substitution se produit au moment où les phyllosilicates se forment. La substitution d’un cation par un autre de valence inférieure mais de taille similaire confère une charge négative nette à la structure du minéral car la charge sur les atomes O environnants n’est pas complètement équilibrée. Par exemple, l’aluminium trivalent (Al3+) peut remplacer certains des atomes de silicium tétravalent (Si4+) dans le feuillet tétraédrique. La charge moindre de Al par rapport à Si crée un déficit de charge qui résulte en une charge négative nette dans le feuillet tétraédrique de la structure. De même, dans les feuillets dioctaédriques, Mg2+, Fe2+ ou d’autres cations divalents peuvent remplacer certains Al3+, produisant une charge négative supplémentaire associée au feuillet octaédrique de la structure. De même, Li+, bien que présent en faible quantité (Tableau 14.1), se substitue aussi parfois au Mg2+ dans les phyllosilicates trioctaédriques (lépidolite).
Pour neutraliser les charges négatives produites par les substitutions isomorphes au sein de la structure des phyllosilicates, des cations de la solution du sol chargés positivement sont attirés à la surface des minéraux. Ces cations neutralisants qui migrent dans la région intercalaire des phyllosilicates sont appelés capacité d'échange cationique (CEC). L’abondance des sites de charges négatives (la densité de charges), l’emplacement des charges négatives (feuillets tétraédriques et/ou octaédriques) et le type de cations neutralisants dans la région intercalaire (par exemple, Na+, Ca2+, Mg2+, K+ ou autre) déterminent la capacité d’expansion du phyllosilicate (argile gonflante) et sa capacité à retenir les éléments nutritifs du sol.
La Région Intercalaire
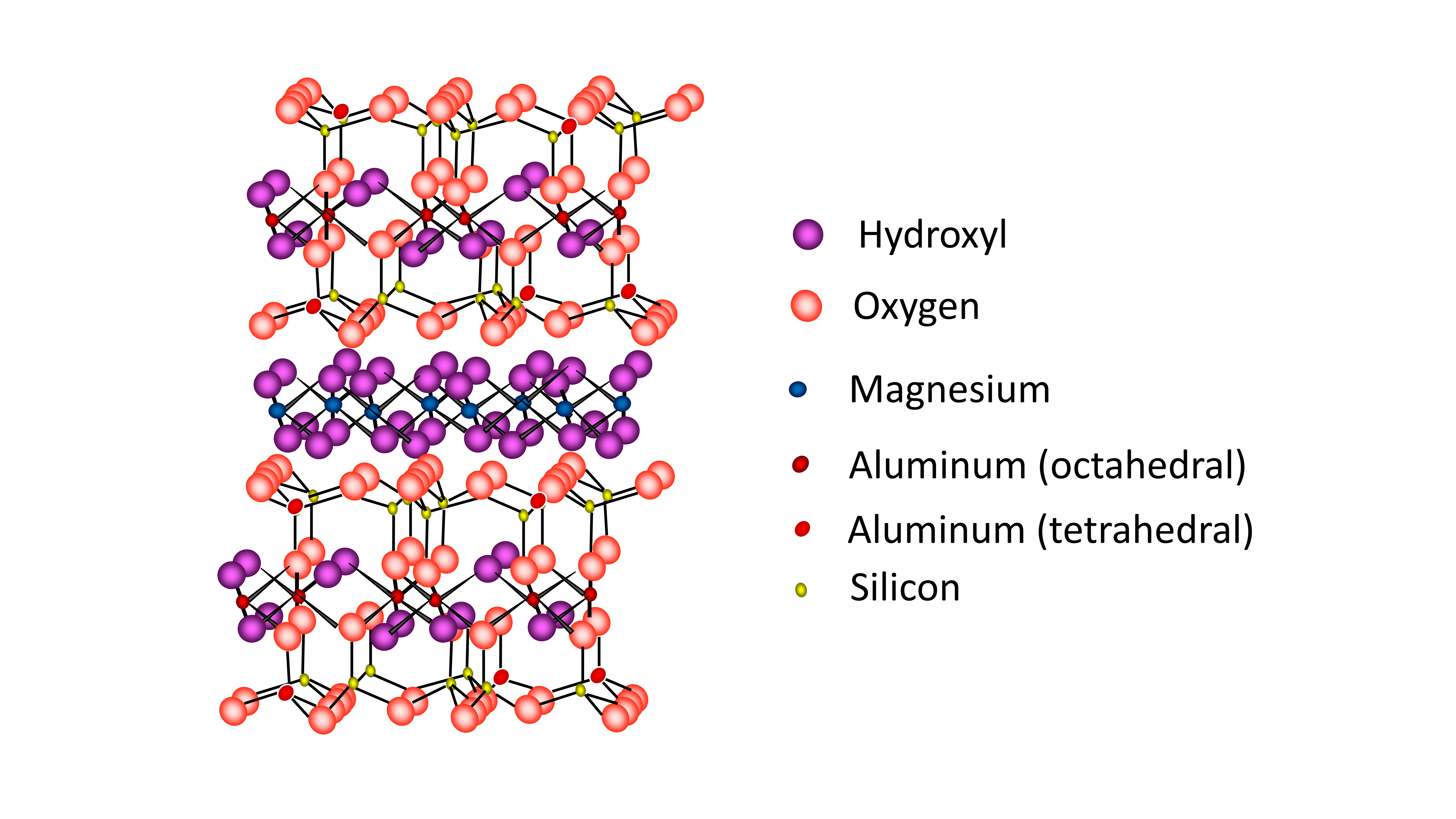
En plus d’attirer et de retenir les cations, la nature de la région intercalaire de certains phyllosilicates peut également être contrôlée par des liaisons hydrogène. Dans les minéraux argileux de type 1:1, les groupes hydroxyles associés au feuillet octaédrique de la structure font face à une couche d’atomes O apicaux de la couche 1:1 adjacente (Figure 14.10). Ainsi, la liaison hydrogène maintient les couches 1:1 étroitement ensemble en une structure très stable. Par conséquent, tous les phyllosilicates de type 1:1 ne se dilatent pas et ne sont pas gonflants.
En revanche, il n’y a pas de liaison hydrogène dans les minéraux argileux 2:1 car les atomes d’O basaux dans les deux couches adjacentes se font face en l’absence de groupes OH– (Figure 14.11). Ainsi, les couches unitaires sont maintenues ensemble par des forces électrostatiques plus faibles. Ces forces électrostatiques faibles résultent de la présence d’un cation dans la région intercalaire qui neutralise les charges négatives causées par les substitutions isomorphes au sein des structures en couches. Les cations intercalaires correspondent à n’importe quelle combinaison de Ca2+, Mg2+, Na+, K+, NH4+, Zn2+, Cu2+, Mn2+, Ni2+, de fer (Fe2+, Fe3+), d’aluminium (Al3+) ou autres. Ces cations sont généralement échangeables et sont associés à une certaine quantité d’eau.
Le degré d’expansion d’un minéral argileux 2:1 dépend de la charge totale de la couche, de l’emplacement des charges (sites tétraédriques face aux sites octaédriques) et de la nature des cations échangeables. Les phyllosilicates 2:1 ayant une faible charge – généralement associés aux feuillets octaédriques – présentent généralement une capacité de gonflement. Les propriétés de gonflement d’un sol sont généralement associées à la présence de smectite (voir Tableau 14.7). Les phyllosilicates 2:1 avec une charge de couche plus élevée (> 0,6 par unité de formule) – généralement associés aux feuillets tétraédriques – sont beaucoup moins susceptibles de présenter des propriétés de gonflement. Les phyllosilicates non-gonflants 2:1 sont les vermiculites et les micas (Tableau 14.7).
L’eau est une molécule polaire, ce qui lui permet le développement de sphères d’hydratation avec le cation échangeable. Les cations divalents tels que Ca2+ et Mg2+ sont généralement associés à deux couches d’eau dans la région intercalaire des minéraux argileux 2:1 dont la charge de couche totale est inférieure. Les gros cations monovalents tels que Na+ et K+, ayant une charge plus faible, n’ont pas le même potentiel que les cations divalents pour maintenir ensemble les structures 2:1; ils permettent donc l’incorporation d’une quantité plus importante de molécules d’eau dans la région intercalaire. Les structures 2:1 peuvent se séparer d’une distance plusieurs fois supérieure à celle de leur configuration d’origine (c’est-à-dire lorsque la structure est « sèche »). De ce fait, on dit que l’argile est une argile gonflante. Ceci est typique du groupe des smectites des phyllosilicates 2:1 (voir ci-dessous). Notez qu’en plus de l’eau, des molécules organiques polaires peuvent également migrer dans la région intercalaire.
Dans certains cas, le cation intercalaire dans les phyllosilicates de type 2:1 peut être hydroxylé six fois (coordination octaédrique). C’est le cas pour Al, Mg et Fe. Dans ce cas, la couche intercalaire se remplit d’un feuillet octaédrique supplémentaire qui ne partage pas d’atomes d’O avec la couche 2:1 principale mais est liée par une liaison hydrogène aux surfaces d’oxydes adjacentes, formant un phyllosilicate de type 2:1:1 (ou 2 :2). Ces types de phyllosilicates appartiennent au groupe des chlorites ou, plus généralement, aux chlorites.
Phyllosilicates communs
Il existe environ 50 espèces de phyllosilicates, mais la plupart sont rares ou se trouvent dans des sols associés à des gisements géologiques spécifiques (par exemple, l’halloysite dans les sols de cendres volcaniques dans les régions humides). Un schéma de classification général des phyllosilicates les plus courants est présenté dans le tableau 14.7. Les cinq espèces de phyllosilicates (kaolinite, smectite [montmorillonite et beidellite], micas [y compris mica hydraté ou illite], vermiculite et chlorite) couramment identifiées dans les sols canadiens sont présentées en détail ci-dessous.
Table 14.7. Classification générale de certains phyllosilicates
| Type | Groupe x = charge par unité de formule |
Sous-groupe | Exemple d’espèces | Formule unitaire idéalisée* |
|---|---|---|---|---|
| 1:1 | Kaolinite x~0 |
Kaolinite | Kaolinite | Si2,Al2O5(OH)4 |
| Halloysite | Si2,Al2O5(OH)4 2H2O | |||
| Serpentines | Serpentine | Si2,Mg3O5(OH)4 | ||
| 2:1 | Pyrophyllite-talc x~0 |
Pyrophyllite | Pyrophyllite | Si4,Al2O10(OH)2 |
| Talc | Talc | Si4,Mg3O10(OH)2 | ||
| Smectite x ≈ 0.25–0.6 |
Dioctaèdre | Montmorillonite | 0.33M+Si4(Al1.67,Mg0.33)O10(OH)2 | |
| Beidellite | 0.33M+(Si3.67Al0.33)(Al2.00)O10(OH)2 | |||
| Nontronite | 0.33M+(Si3.67Al0.33)(Fe(III)2.00)O10(OH)2 | |||
| Trioctaédrique | Saponite | 0.33M+(Si3.67Al0.33)(Mg3.00)O10(OH)2 | ||
| Hectorite | ||||
| Vermiculite x ≈ 0.6–0.9 |
Dioctaédrique | Vermiculite dioctaédrique | 0.86M+(Si3.47Al0.53)(Al1.67Mg0.33)O10(OH)2 | |
| Trioctaédrique | Vermiculite trioctaédrique | 0.86M+(Si3.47Al0.53)(Al1.67Mg0.33)O10(OH)2 | ||
| Micas x ≈ 1 |
Dioctaédrique | Muscovite | K1.00(Si3.00Al1.00)(Al2.00)O10(OH)2 | |
| Paragonite | ||||
| Trioctaédrique | Biotite | K1.00(Si3.00Al1.00)(Fe(II)3.00)O10(OH)2 | ||
| Phlogopite | ||||
| 2:1:1 | Chlorite x = variable |
Di-Dioctaédrique | Donbassite | (Al2(OH)6)(Si,Al)4Al2O10(OH)2 |
| Tri-Dioctaédrique | Sudoite | (Mg3(OH)6)(Si,Al)4 Al2O10(OH)2 | ||
| Di-Trioctaédrique | Ferri-sudoite | (Al2(OH)6)(Si,Al)4(Mg,Fe)3O10(OH)2 | ||
| Tri-Trioctaédrique | Clinochlore | (Mg3(OH)6)(Si,Al)4(Mg,Fe)3O10(OH)2 | ||
| *La séquence d'éléments dans l'unité de formule idéalisée suit l'ordre : cation échangeable (le cas échéant) ou feuillet octaédrique intercalaire ; cations tétraédriques; cations octaédriques; oxygènes et hydroxyles. Dans chaque feuillet, le cation majeur (par exemple la silice) est suivi du ou des cations de substitution isomorphe. | ||||
La kaolinite est un minéral argileux de type 1:1 constitué d’un feuillet tétraédrique et d’un feuillet octaédrique (Figure 14.10). Les couches 1:1 sont maintenues ensemble par des liaisons hydrogène intercalaires. En conséquence, la structure n’est pas extensible car les molécules d’eau ne peuvent pas pénétrer dans la région intercalaire. La kaolinite gonfle très peu lorsqu’elle est mouillée et se rétracte peu lorsqu’elle est sèche. La couche intercalaire n’est pas exposée, ce qui réduit la surface spécifique. Le minéral n’a presque pas de substitution isomorphe; par conséquent, les minéraux de kaolinite ont une faible charge de surface et une faible CEC, bien qu’ils aient quelques sites de charge variable en bordure. La kaolinite est le phyllosilicate de type 1:1 le plus courant et est omniprésente dans les sols canadiens.

La smectite est un phyllosilicate de type 2:1 à forte capacité d’expansion (Figure 14.11). Il existe deux espèces communes de smectite : la montmorillonite et la beidellite. La quantité de substitution isomorphe dans les deux minéraux est modérée par rapport aux autres phyllosilicates de type 2:1. La charge de couche pour les smectites est de l’ordre d’environ 0,25 à 0,6 site par unité de formule (Mx+(Si,Al)4(Al,Mg)2O10(OH)2) où Mx+ fait généralement référence à des cations échangeables (par exemple, Na+, Mg2+, K+, Ca2+, etc.). La montmorillonite et la beidellite ont des charges de couche totales comparables mais se différencient par l’emplacement des charges de couche dans la structure (Tableau 14.7). Dans la montmorillonite, la majeure partie de la charge de couche est logée dans le feuillet octaédrique tandis que pour la beidellite, la majeure partie de la charge est associée au feuillet tétraédrique. Leur surface spécifique est élevée (environ 800 m2 g-1) et les molécules d’eau et les cations peuvent pénétrer dans l’espace intercalaire.
Les sols riches en smectite sont collants lorsqu’ils sont humides et forment de grandes fissures dans des conditions sèches. Les sols à texture argileuse contenant plus de 60 % d’argile, dont au moins la moitié est de la smectite, sont classifiés comme des Vertisols au Canada (SCGW, 1998). Ces sols sont décrits comme des sols « d’auto-paillage » car des cycles répétés d’humectation et de dessiccation forment des faces de glissement. Ces dernières sont des surfaces polies au contact de peds opposés. Les peds se trouvent le long de fissures causées par des mouvements de matériaux lors de la rétractation et de la dilatation en réponse aux cycles d’humectation et de dessiccation. Le processus pédogénétique à l’origine de la formation de faces de glissement est appelé argilipédoturbation, lequel est particulièrement courant dans les sols vertisoliques qui se forment à partir de matériaux parentaux glacio-lacustres riches en smectite reytrouvés généralement dans les provinces des Prairies canadiennes.

Les micas, y compris le mica/illite hydraté, sont des minéraux argileux de type 2:1 avec deux feuillets tétraédriques prenant en sandwich un feuillet octaédrique. Les couches 2:1 sont maintenues étroitement ensemble par un ion K+ (de grand rayon) dans la région intercalaire (Figure 14.12). La structure des micas est similaire à celle des smectites sauf que la charge de la couche est beaucoup plus élevée (environ 1 par unité de formule) et les substitutions isomorphes se produisent presque exclusivement dans les feuillets tétraédriques. Le potassium occupe presque exclusivement la région intercalaire. La charge de couche très élevée, combinée à l’emplacement de la charge dans les feuillets tétraédriques très près de la région intercalaire, empêche l’expansion du minéral. Les micas sont des minéraux primaires dérivés du granite et des roches métamorphiques du Bouclier Canadien. Des mica/illites hydratés se forment dans le sol à partir de l’altération partielle des micas. Les micas et le mica/illite hydraté se trouvent dans presque tous les sols du Canada. Il existe deux espèces communes de micas qui se distinguent par la nature du feuillet octaédrique, à savoir la muscovite dioctaédrique et la biotite trioctaédrique (Tableau 14.7).
Le mica/illite hydraté est un produit d’altération partielle des micas, dont une partie (jusqu’à 20 à 30 %) des ions K+ de la couche intercalaire a été éliminée et remplacée par d’autres cations échangeables. Des ions Al3+ tétraédriques sont aussi absents de la structure. Le mica/illite hydraté est généralement produit par l’altération de la muscovite dioctaédrique, tandis que la structure de la biotite trioctaédrique se décompose généralement en totalité en raison de la relative vulnérabilité aux attaques chimiques du fer (Fe(II)) de la structure.
Bien que la structure de base des micas et du mica/illite hydraté soit similaire à celle de la smectite, leurs propriétés sont très différentes, principalement en raison de la plus grande quantité de substitution isomorphe dans le feuillet tétraédrique, où Al3+ remplace Si4+ (Figure 14.12). La forte charge négative située dans les feuillets tétraédriques près de la surface intercalaire est équilibrée par des ions K+, bloquant les couches adjacentes ensemble. Ainsi, le minéral a une expansion limitée car les molécules d’eau ne peuvent pas pénétrer dans l’espace intercalaire. Puisque le piégeage des ions K+ entre les couches adjacentes empêche l’expansion, la CEC est faible.

La vermiculite est un phyllosilicate de type 2:1 dont la structure est similaire à celle de la smectite et des micas. La charge de la couche est comprise entre 0,6 et 0,9 site par unité de formule (Mx+(Si,Al)4Al2O10(OH)2),
ce qui est intermédiaire entre les micas et la smectite (Figure 14.13). Comme dans les micas, la charge de couche est située presque exclusivement à l’intérieur du feuillet tétraédrique. La capacité d’expansion de la vermiculite est limitée et ses capacités de gonflement et de rétraction sont modérées. Une charge de couche inférieure à celle des micas mais supérieure à celle de la smectite combinée à des substitutions isomorphes majoritairement localisées dans le feuillet tétraédrique, confère à la vermiculite une charge de surface et une CEC élevées. Du fait de sa structure similaire à celle des micas, la vermiculite est capable de piéger les ions K+ dans la région intercalaire.
La chlorite est le nom d’un minéral, ainsi que d’un groupe de phyllosilicates de type 2:1:1 (ou 2:2) constitués d’une structure en couches 2:1 avec une couche d’hydroxydes dans la région intercalaire (Kohut et Warren 2002). La chlorite minérale est l’espèce la plus commune au sein du groupe des chlorites. La structure est composée d’une couche d’hydroxyde d’Al, de Mg ou de Fe en coordination sextuple (octaédrique) dans la région intercalaire entre les couches de type 2:1 (Figure 14.13). Cette couche hydroxyle intercalaire ne partage pas d’atomes O avec la couche principale 2:1. La couche d’hydroxyde est liée aux couches 2:1 adjacentes par une liaison hydrogène et n’est pas expansible (Figure 14.13). La quantité de substitutions isomorphe est variable, mais comme la structure n’est pas expansible, la CEC globale est faible.
Capacité d’Échange et Surface Spécifique
La fraction argileuse de la plupart des sols canadiens contient généralement un mélange des cinq phyllosilicates courants ainsi que de petites quantités d’aluminosilicatesamorphes et d’oxyhydroxydes de Fe et/ou d’Al. Les proportions sont principalement fonction de l’origine des matériaux d’origine du sol. Les variations des propriétés globales d’échange d’ions et d’adsorption des sols dépendent du mélange de phyllosilicates en présence. Les deux principales différences entre les phyllosilicates, c’est-à-dire leur CEC et leur surface spécifique, sont résumées dans le tableau 14.8. La kaolinite a très peu de substitutions isomorphes et n’est pas expansible. Par conséquent, elle a une CEC très faible et toute la surface réactive est associée aux surfaces externes de la structure. Le mica/illite hydraté et les chlorites ont une quantité significative de substitutions isomorphes. Cependant, comme leurs régions intercalaires contiennent respectivement des K+ fermement liés et une couche d’hydroxyde supplémentaire, l’expansion de la couche est inhibée. Ainsi, la CEC est limitée et les structures ne sont pas expansibles. Les smectites ont un nombre de substitutions isomorphes intermédiaire couplé à la capacité d’accueillir des cations échangeables dans la région intercalaire. Par conséquent, la CEC et la superficie totale sont élevées. Les sols contenant des smectites sont connus pour leur forte plasticité, leur cohésion et leur capacité caractéristique à gonfler lorsqu’ils sont humides et à rétrécir lorsqu’ils sont secs. Les vermiculites ont davantage de substitutions isomorphes par rapport aux smectites, et ont donc une CEC plus élevée; cependant, la plupart de la charge dans les vermiculites est concentrée dans les feuillets tétraédriques proches de la région intercalaire, ce qui lui confère une plus grande attraction électrostatique pour les cations échangeables de la région intercalaire. En conséquence, bien que la surface spécifique totale de la vermiculite soit similaire à celle des smectites, l’expansion de la couche des structures de vermiculite est limitée en raison de l’entrée restreinte de l’eau dans la région intercalaire.
Table 14.8. Gammes de Capacité d’Échange Cationique (CEC), et surfaces internes et externes dans des phyllosilicates et autres colloïdes communs. (Adapté de Weil et Brady, 2017)
| Phyllosilicate/Colloïde | CEC (cmol(+) kg-1) |
Surface spécifique externe (m2 g-1) |
Surface spécifique interne (m2 g-1) |
|---|---|---|---|
| Kaolinite | 2-16 | 5-15 | ~0 |
| Chlorite | 10 - 40 | 5-20 | ~0 |
| Mica/Illite hydraté | 20 - 40 | 100 - 125 | ~0 |
| Smectite | 60 - 120 | 80 - 140 | 570 - 660 |
| Vermiculite | 100 - 150 | 70 - 120 | 600 - 700 |
| Aluminosilicates amorphes | 5 - 350 | 500 - 1450 | ~0 |
| Fe/Al | ~0 - 3 | 200 - 500 | ~0 |
| oxyhydroxides |
Pouvez-vous creuser!
Bien que cela ne soit pas confirmé, certaines données suggèrent que les phyllosilicates pourraient avoir été un catalyseur important à l’origine de la vie sur notre planète (Berman, 2019). Les minéraux argileux agissent comme des catalyseurs très efficaces dans la polymérisation des acides aminés et des nucléotides. L’ARN adsorbé sur des minéraux argileux peut être encapsulé dans des vésicules. Une fois formées, ces vésicules pourraient croître et se diviser en incorporant des acides gras, assurant ainsi la médiation de la réplication des vésicules à travers des cycles de croissance et de division (Brack, 2013)
DISTRIBUTION DES PHYLLOSILICATES DANS LES SOLS CANADIENS
La répartition des phyllosilicates dans les sols canadiens varie selon la région physiographique (Kodama, 1979); la plupart des phyllosilicates sont hérités des matériaux parentaux déposés par le retrait des glaciers continentaux à la fin du Pléistocène. Par exemple, certains minéraux argileux comme la kaolinite, le mica (c’est-à-dire la biotite et la muscovite) et la chlorite sont hérités directement du Bouclier canadien. Dans la région de la Cordillère et des plaines intérieures, d’autres minéraux argileux sont dérivés de la dégradation glaciaire des sédiments de la période du Crétacé (il y a 65 millions d’années). La comparaison des données pour les fractions argileuses des sols, par exemple entre celles des plaines intérieures de l’ouest et celles trouvées dans l’est du Canada (basses terres du Saint-Laurent), montre une différence marquée dans la suite de minéraux argileux (Tableau 14.9). La région des plaines intérieures de l’ouest du Canada contient généralement de la smectite, du mica de taille argileuse, de la chlorite et un peu de kaolinite (Kodama, 1979; Dudas et Pawluk, 1982; Spiers et al. 1989a; Spiers et al. 1989b; Warren et Dudas, 1992). En revanche, la fraction argileuse des sols des basses terres de l’ouest du Saint-Laurent (Ontario) provient de sédiments beaucoup plus anciens formés au cours du Dévonien (359 millions) et du Silurien (443 millions) et est dominée par le mica, la chlorite, la vermiculite et la kaolinite avec quelques traces de smectite (Kodama, 1979). Le matériel parental a reçu des contributions du Bouclier canadien dans les deux régions. Il n’y a que très peu d’évidence d’altération dans les divers assemblages de minéraux argileux des diverses régions physiographiques, et la corrélation entre les types génériques de sol est négligeable. Les sols podzoliques et, dans une moindre mesure, les sols brunisoliques et gleysoliques où les minéraux de chlorite hérités semblent s’altérer en phyllosilicates expansibles, font exception (Kodama, 1979).
Table 14.9. Gamme d’abondance des phyllosilicates typiquement trouvés dans la fraction argileuse des sols des plaines intérieures et de l’ouest des basses terres du St-Laurent
| Région des plaines intérieures | Basses terres du St-Laurent | |||
|---|---|---|---|---|
| Phyllosilicate | g kg-1 | Phyllosilicate | g kg-1 | |
| Kaolinite | 70-110 | Kaolinite | 120-150 | |
| Muscovite distincte | 250-340 | Muscovite distincte | 350-450 | |
| Montmorillonite | 470-490 | Vermiculite | 180-220 | |
| Chlorite | 120-160 | Chlorite | 240-300 | |
| Vermiculite | trace | Smectite | trace | |
Aluminosilicates Amorphes
D’autres aluminosilicates hydratés se trouvent également dans le sol mais ceux-ci sont structurellement différents des phyllosilicates. Ils sont décrits comme amorphes (sans forme) ou nanocristallins et contrairement aux phyllosilicates n’ont pas de structure cristalline ordonnée précise. L’allophane et l’imogolite sont des exemples de minéraux aluminosilicatés amorphes, couramment formés lors d’une altération rapide dans les sols de cendres volcaniques trouvés dans les climats humides de pays comme la Nouvelle-Zélande, le Chili et le Japon. Les cendres volcaniques s’altèrent rapidement et se dissolvent généralement en libérant de grandes quantités d’aluminium et de silice solubles, qui peuvent précipiter sous forme d’allophane ou d’imogolite. Les aluminosilicates amorphes ne sont pas expansibles, mais sont extrêmement petits (<4 nm de diamètre) et ont une surface spécifique (700-1500 m2 g-1) et une capacité d’échange d’ions très élevées. Bien qu’on les trouve essentiellement dans les sols de cendres volcaniques, ils peuvent être présents en petites quantités dans certains sols aux côtés des phyllosilicates. La présence d’imogolite, par exemple, est documentée dans certains sols podzoliques humo-ferriques de Colombie-Britannique (Grand et Lavkulich, 2013; 2015), ailleurs au Canada (McKeague et Kodama, 1981; Wang et al., 1991) et dans les sols solonetziques Solodisés en Alberta (Spiers et al., 1984).
OXYHYDROXYDES DE FER ET D’ALUMINIUM
Les argiles oxyhydroxydes (sesquioxydes) sont des oxydes secondaires, des hydroxydes et des minéraux d’oxydes hydratés de Fe, d’Al et de Mn. Certains sont bien cristallisés (par exemple, la gibbsite, l’hématite, la goethite), tandis que d’autres sont amorphes. Ces minéraux sont habituellement responsables des teintes rouges, jaunes, brunes et bleuâtres observées dans les sols. Les argiles d’oxydes cristallines sont composées de feuillets de groupes O et/ou hydroxyle dans un arrangement octaédrique avec Fe, Al ou Mn. Elles n’ont pas de feuillets tétraédriques de Si dans leurs structures. Les structures octaédriques de ces minéraux sont maintenues ensemble par une liaison hydrogène; elles ne se dilatent pas. Les oxydes et hydroxydes de fer autrefois appelés « limonite » se sont révélés plus tard être en fait un mélange de minéraux d’oxyhydroxyde de fer.
Ces minéraux ne portent pas de charges négatives permanentes identifiables, mais ont une forte charge de surface associée à des groupes hydroxyles de surface. La capacité d’échange des oxyhydroxydes dépend fortement du pH. À faible pH, la protonation des groupes hydroxyles produit des charges positives. À pH élevé, la dissociation des groupes hydroxyles produit des charges négatives. Les minéraux oxyhydroxylés sont qualifiés d’amphotères (peuvent agir comme une base ou un acide) du fait de leur CEC variable. Ces minéraux ont généralement une CEC nette à pH élevé, et une capacité d’échange anionique nette à pH faible.
Les oxyhydroxydes se trouvent généralement en petites quantités dans les sols canadiens (Grand et Lavkulich, 2013; 2015; Spiers et al., 1984). Ils sont très réactifs en raison de leur surface spécifique très élevée. La présence de ces minéraux influence de manière significative certaines propriétés du sol telles que l’adsorption et l’échange des ions. Par exemple, les oxyhydroxydes de Fe sont capables d’adsorber et de retenir de grandes quantités d’ions phosphate, molybdate et borate de la solution du sol, ainsi que de piéger des métaux tels que Cu, Pb, Zn, Co, Cr et Ni. Certains oxyhydroxydes de Fe courants sont répertoriés dans le tableau 14.10.
Table 14.10. Quelques-uns des oxyhydroxydes secondaires communément trouvés dans le sol
| Minéral | Formule | Remarques |
|---|---|---|
| Goethite | α-FeOOH | Le plus commun et abondant oxyhydroxyde des sols. Jaune à ocre. Granulométrie très fine. Précipite très vite à partir du Fe3+ en solution. Très petits cristaux(<0.5 um) aciculaires. |
| Lepidocrocite | γ-FeOOH | Trouvé dans les sols mal drainés sous forme de monticules orange vif mais PEU commun. Formé par l’oxydation de précipités de Fe(II)(OH)2 en absence de CO2 à pH 5-7. Cristaux lamellaires extrêmement petits (0.1-0.5 um). |
| Hématite | α-Fe2O3 | Deuxième en abondance après la goethite. Prévalent dans les sols tropicaux. Se forme par déshydratation de la goethite et de la ferrihydrite. De très petites quantités confèrent au sol une couleur rouge. |
| Ferrihydrite (ou hydroxyde de fer hydraté) |
(Fe(III))2O3·0.5H2O mais aussi 5Fe2O3·9H2O |
Composition très variable. Précipite directement à partir d'eau oxygénée riche en Fe ou en lien avec l’activité biologique de bactéries. Rouge-orange. Forme des nanocristaux. Métastable. |
CARBONATES
Certains minéraux carbonatés communément trouvés dans les sols sont présentés dans le tableau 14.11. La majorité des carbonates du sol proviennent de sources primaires, c’est-à-dire de roches sédimentaires, alors que certains sont des précipités secondaires, généralement trouvés dans les horizons souterrains. La structure cristalline de nombreux minéraux carbonatés reflète la symétrie trigonale de l’ion carbonate qui se forme généralement en combinaison avec Ca, Na, U, Fe, Al, Mn, Ba, Zn, Cu, Pb et d’autres éléments. Il existe environ 80 minéraux carbonatés connus; cependant, les minéraux carbonatés de loin les plus courants dans les sols canadiens sont la calcite (CaCO3) et la dolomite ([Ca,Mg]CO3) provenant principalement du substratum rocheux datant du Paléozoïque. La plupart des minéraux carbonatés contiennent généralement des traces d’autres éléments liés au carbonate (voir ci-dessus) dans leurs structures.
La calcite est peu soluble, mais la solubilité augmente à bas pH; elle ne se trouve généralement pas dans les sols dont le pH est inférieur à 6,0. La calcite présente dans le solum se dissout généralement et est lessivée avant de se redéposer dans le profil inférieur du sol, parfois sous forme de nodules de calcite secondaires (Miller et al., 1987; Wang et Anderson, 2000). L’aragonite et les autres minéraux associés au carbonate de calcium ne sont pas communs dans les sols en raison de leur faible stabilité.
Table 14.11. Quelques-uns des carbonates les plus communs
| Minéral | Formule idéalisée | Remarques |
|---|---|---|
| Calcite* | CaCO3 | Composant minéral principal du calcaire. Typiquement instable et se dissout dans le solum supérieur mais peut précipiter dans l'horizon C, généralement sous des régimes climatiques subhumides ou plus secs |
| Aragonite* | CaCO3 | Issu des coquillages fossilisés d’origine géologique. Instable et se dissout dans le sol |
| Dolomite | (Ca,Mg)CO3 | Issu de roches sédimentaires (dolomie) d’origine géologique. Instable et se dissout dans le sol |
| Sidérite | Fe(II)CO3 | Issu de roches sédimentaires ou métamorphiques. Instable dans le sol mais dissolution dans le sol très lente |
| Magnésite | MgCO3 | Très soluble dans le sol |
| Trona | Na2CO3 | Très soluble dans le sol. Trouvé le plus souvent dans les sols Solonetziques à pH élevé (~9.5) |
| *La calcite et l’aragonite sont polymorphes. Ils ont des formules chimiques similaires mais des structures crystallines différente. | ||
La dolomite (Ca,Mg(CO3)2) est un minéral de carbonate de calcium-magnésium communément trouvé dans les sols dérivés d’un substratum rocheux de dolomie. Comme la calcite, la dolomite se dissout dans le profil supérieur, apportant du Ca lors de la formation de calcite secondaire par précipitation dans le profil inférieur; le composant de magnésium est quant à lui principalement lessivé ou absorbé par les racines des plantes. Une partie du Mg2+ peut se substituer au Ca2+ dans les carbonates secondaires si la lixiviation est limitée, mais la quantité est mineure (<10%) (St. Arnaud, 1979).
Les carbonates confèrent un certain nombre de propriétés au sol, notamment :
- le maintien d’une saturation en bases élevée;
- le maintien d’une quantité de Ca et de Mg échangeables élevée;
- le maintien d’un pH >7 ;
- l’adsorption/précipitation des ions PO43-;
- l’adsorption/précipitation de métaux (par exemple, Al, Zn, Pb, Fe, Ni, Mn); et,
- la stabilisation des argiles et de la structure du sol.
MINÉRAUX PHOSPHATÉS
Minéraux Phosphatés Primaires
L’abondance moyenne du P dans la croûte terrestre est de 1050 mg kg-1, tandis que la teneur totale en P dans les sols est généralement comprise entre 400 et 800 mg kg-1. Le phosphate peut facilement se combiner avec environ 30 éléments différents pour former plus de 300 minéraux phosphatés (Bleam, 2017). Certains sont des minéraux phosphatés primaires dérivés de roches sédimentaires et ignées, mais la plupart sont des minéraux contenant du phosphate sous forme de PO4 substitué dans leurs structures, ce qui est considéré comme une « impureté ».
Les minéraux phosphatés primaires « purs » les plus courants appartiennent au groupe « apatite » et ont la formule chimique générale : Ca5(PO4)3(F,Cl,OH).
L’apatite se présente généralement sous forme de très petits cristaux hexagonaux avec une variété de couleurs, notamment le vert, le vert bleuâtre, le brun, le blanc, le jaune, le rouge, le gris et autres selon la présence d’impuretés. Les noms spécifiques des minéraux contenant les trois membres terminaux les plus communs de l’apatite sont les suivants :
- fluoroapatite Ca5(PO4)3F
- chloroapatite Ca5(PO4)3Cl
- hydroxyapatite Ca5(PO4)3OH
Dans la nature, ces minéraux sont mélangés, mais l’un des trois composants (F, Cl et OH) domine dépendamment de la source. Le phosphate rocheux ou la phosphorite contient généralement de fortes concentrations en ces minéraux et est extrait principalement pour la production d’engrais phosphatés. Dans les sols, l’apatite peut provenir de presque toutes les roches ignées ou métamorphiques présentes à l’état de phase accessoire (Nezat et al., 2007) mais rarement à l’état de phase majeure dans la roche. La structure minérale de l’apatite peut également contenir des substitutions avec une grande variété d’atomes, notamment le carbonate (CO32-), l’arséniate (AsO42-), le V, le Th, le Pb, le Sr, le Ba, l’U et les éléments de terres rares.
Minéraux Phosphatés Secondaires
En général, le phosphate dans le sol (dérivé de sources minérales primaires et les engrais phosphatés) réagit facilement avec Ca2+, Al3+, and Fe2+ and Fe3+ en formant une variété de précipités secondaires (Tableau 14.12). Les phosphates de fer (vivianite et strengite) sont plus courants dans les sols suboxiques où le fer dissous est plus abondant. Les phosphates d’aluminium (varisite) sont le plus souvent associés aux sols acides où le phosphate précipite d’abord sous la forme d’un intermédiaire amorphe qui finit par se transformer en varisite (Lindsay et al., 1959). Les phosphates de calcium secondaires sont généralement présents dans les sols calcaires contenant de la calcite minérale comme source de calcium (Kar et al., 2012). La brushite secondaire se trouve généralement sous forme de précipité dans les sols calcaires en raison des fortes concentrations d’engrais qui sont apportés pour la production agricole (McLaughlin et al., 2011).
Table 14.12. Quelques-uns des minéraux phosphatés secondaires les plus communément trouvés dans le sol
| Groupe | Nom du minéral | Formule idéalisée | Présence |
|---|---|---|---|
| Fer | Vivianite | Fe(II)3(PO4)·2.8H2O | Sols humides/inondés |
| Phosphates | Strengite | Fe(III)(OH)2(H2PO4) or FePO2·2H2O | Sols humides/inondés |
| Phosphates d’aluminium | Variscite | Al(OH)2(H2PO4) or AlPO4·2H2O | Sols acides |
| Phosphates de calcium | Brushite | CaHPO4·2H2O | Sols calcaires |
SULFATES ET SULFIDES
Les minéraux sulfurés sont absents dans les sols bien à imparfaitement drainés, mais peuvent être présents dans certains sols Glgleysoliques ou organiquesmal drainés. La pyrite minérale sulfurée (FeS2) est un minéral primaire très commun dans les déchets miniers, mais peut aussi se trouver dans les schistes et sous forme de précipité dans certains sols de marée en réponse à des conditions fortement réductrices. La pyrite, une fois exposée à l’atmosphère, s’oxyde facilement en libérant du fer et de l’acide sulfurique :
2FeS2 + 7O2 + 2H2O → 2Fe2+ + 4H+ + 4SO42-
Au fur et à mesure que le fer réduit (Fe(II)) dans la structure sulfurée est libéré, il s’oxyde et précipite sous forme d’oxyhydroxyde. Le S oxydé (SO42-) se combine ensuite avec l’eau pour produire un acide (H2SO4). L’exposition anthropique de minéraux sulfurés lors d’opérations minières ou d’autres excavations entraîne un drainage minier acide (DMA). Des valeurs de pH du sol inférieures à 2,0 peuvent résulter de l’oxydation des sulfures. Le sulfate dérivé de l’oxydation de sulfures ou d’autres sources produit du gypse (Tableau 14.13), généralement en réponse à un pH élevé et à la présence de Ca soluble.
Table 14.13. Quelques-uns des minéraux soufrés
| Type | Minéral | Formule idéalisée |
|---|---|---|
| Sulfures | Pyrite | FeS2 |
| Pyrrhotite | Fe(1-x)S (x = 0 to 0.2) | |
| Sphalérite | ZnS | |
| Galèna | PbS | |
| Chalcopyrite | CuFeS2 | |
| Arsénopyrite | FeAsS | |
| Pentlandite | (Fe,Ni)9S8 | |
| Cinabre | HgS | |
| Marcasite | FeS2 | |
| Sulfates | Gypse | CaSO4·2H2O |
| Barytine | BaSO4 |
RÉSUMÉ
En résumé, le matériau parental de la plupart des sols minéraux du Canada a été déposé par l’action physique des glaciers. La plupart des minéraux de ces sols sont dérivés soit du Bouclier canadien, soit du remaniement et des dépôts d’autres sources au cours des glaciations antérieures à la dernière glaciation du Wisconsinien. Les minéraux primaires qui composent la majorité de la fraction sableuse des sols canadiens sont généralement dominés par le quartz et les feldspaths, avec une petite quantité de minéraux lourds. L’altération chimique des sables contribue à la formation de minéraux secondaires et d’éléments nutritifs des plantes. Les phyllosilicates dominent la fraction argileuse et constituent la phase inorganique la plus chimiquement active du sol. Les différences de composition en phyllosilicates de la fraction argileuse des régions de l’ouest et de l’est du Canada sont principalement dues à l’origine des matériaux parentaux. Les limons, dont la taille est intermédiaire entre les fractions sableuse et argileuse, sont constitués d’un mélange physique de minéraux provenant de ces deux fractions.
RÉFÉRENCES
Ando, S. 2020. Gravimetric separation of heavy minerals in sediments and rocks. Minerals. 10(3): 273
Berman, J.J. 2019. Evolutions clinical guidebook. Translating ancient genes into precision medicine. Academic Press. 357pp.
Beutelspacher, H., and H. Van Der Marel, 1968. Atlas of electron microscopy of clay minerals and their admixtures. A picture atlas. Elsevier. 333pp.
Bleam, W.F. 2017. Soil and environmental chemistry, 2nd Edition. Academic Press. 573pp.
Brack, A. 2013. Clay minerals and the origin of life. Chapter 10.4. Developments in Clay Science. Elsevier. 507-521pp.
Bowen, H J M, 1979. Environmental chemistry of the elements. Academic Press, London, New York.
Cousineau, P.A. 2020. Personal Communication.
Dudas, M.J. and Pawluk, S. 1982. Re-evaluation of the occurrence of interstratified and other phyllosilicates in southern Alberta soils. Canadian Journal of Soil Science. 62: 61-69.
Fanning, D., Rabenhorst, M. and R. Fitzpatrick. 2017. Historical developments in the understanding of acid sulfate soils. Geoderma 308: 191-206. http://dx.doi.org/10.1016/j.geoderma.2017.07.006
Finkl C.W. 1981. Soil mineralogy. In: Mineralogy. Encyclopedia of Earth Science. Springer, Boston, MA. https://doi.org/10.1007/0-387-30720-6_133
Grand, S and L.M. Lavkulich. 2013. Potential influence of poorly crystalline minerals on soil chemistry in Podzols of southwestern Canada. European Journal of Soil Science 64(5):651-660.
Grand, S and L.M. Lavkulich. 2015. Short-range order mineral phases control the distribution of important macronutrients in coarse-textured forest soils of coastal British Columbia, Canada. Plant and Soil. DOI 10.1007/s11104-014-2372-6
Havlin, J. L, Tisdale S.L, Nelson W.L., Beaton, J.D. 2005. Soil fertility and fertilizers: An introduction to nutrient management, 7th edition. Pearson. 528pp.
Kar, G., Peak, D. and J. Schoenau. 2012. Spatial Distribution and Chemical Speciation of Soil Phosphorus in a Band Application. Soil Science Society of America Journal 76(6):2297-2306. DOI: 10.2136/sssaj2012.0146
King, H.M., 2020. Feldspar. Geology.com. https://geology.com/minerals/feldspar.shtml
Kodama, H. 1979. Clay minerals in Canadian soils; their origin, distribution and alteration. Canadian Journal of Soil Science. 59: 37-58.
Kohut, C.K. and Warren, C.J., 2002. Chlorites. In Amonette, J.E. Bleam, W.F. Shultz, D.G. and J.B Dixon (eds.). Soil Mineralogy with Environmental Applications. Soil Science Society of America Book Series. No. 7, Madison, WI. 531-553pp.
Lindsay, W.L. Peech, M. Clark, J.S. 1959. Solubility criteria for the existence of variscite in soils. Soil Science Society of America Journal. 23(5): 357-360.
Loganathan, P. 1987. Soil Quality, Physical properties of the soil. http://www.fao.org/3/AC172E/AC172E04.htm#ch4
Liu, C. Li, X. Xu, F. Huang, P.M. 2003. Atomic force microscopy of soil inorganic colloids. Soil Science and Plant Nutrition. 49(1): 17-23.
Maitre J., Bouchard K., Bédard L.P. 2019. Mineral grains recognition using computer vision and machine learning. Computers & Geosciences 130: 84-93.
McKeague, J.A. and H. Kodama. 1981. Imogolite in cemented horizons of some British Columbia soils. Geoderma 25(3-4): 189-197.
McLaughlin, M.J., McBeath, T.M., Smernik, R., Stacey, S.P., Ajiboye, B. and G. Guppy. 2011. The chemical nature of P accumulation in agricultural soils—implications for fertiliser management and design: an Australian perspective. Plant Soil 349:69–87.
Miller, J. J., Dudas, M. J. and Longstaffe, F. J. 1987. Identification of pedogenic carbonate minerals using stable carbon and oxygen isotopes, X-ray diffraction and SEM analyses. Canadian Journal of Soil Science. 67: 953-958.
Mindat. 2020. https://www.mindat.org/
Mineralogical Society of America (MSA) 1997-2020. http://www.minsocam.org/msa/collectors_corner/faq/faqmingen.htm
Mineralogical Society of America (MSA) 2004-2020. http://www.handbookofmineralogy.org/search.html?p=all
Nezat, C.A. Blum, J.D. Yanai, R.D. and Hamburg, S.P. 2007. A sequential extraction to determine the distribution of apatite in granitoid soil mineral pools with application to weathering at the Hubbard Brook Experimental Forest, NH, USA. Applied Geochemistry. 22(11): 2406-2421.
Parikh, S. J. & James, B. R. 2012. Soil: The foundation of agriculture. Nature Education Knowledge. 3(10): 2
Pauling, L. 1929. The principles determining the structure of complex ionic crystals. Journal of the American Chemical Society. 51(4): 1010-1026.
Rutter, N.W. and White, M. 2015. Glaciation. The Canadian Encyclopedia. https://www.thecanadianencyclopedia.ca/en/article/glaciation
Smart, P. and Tovey, N.K. 1981. Electron microscopy of soils and sediments: Examples. Oxford University Press. 400pp.
Soil Classification Working Group (SCWG), 1998. The Canadian system of soil classification. 3rd Edition. Agriculture and Agri-Food Canada. Publication 1646. 187pp.
Spiers, G.A., M.J. Dudas and L.W. Turchenek. 1989a. The chemical and mineralogical composition of soil parent materials in northeast Alberta. Canadian Journal of Soil Science. 69: 721-738.
Spiers, G.A., M.J. Dudas, K Muehlenbachs and L.W. Turchenek. 1989b. Mineralogy and oxygen isotope geochemistry of clays from surficial deposits in the Athabasca Tar Sands area. Canadian Journal of Earth Sciences. 21: 53-60.
Spiers, G.A., Pawluk, S., M.J. Dudas. 1984. Authigenic mineral formation by Solodization. Canadian Journal of Soil Science. 64: 515 532.
St. Arnaud, R.J. 1979. Nature and distribution of secondary soil carbonates within landscapes in relation to soluble Mg++/Ca++ ratios. Canadian Journal of Soil Science. 59: 87-98.
Wang, D. and Anderson, D.W. 2000. Pedogenic carbonate in Chernozemic soils and landscapes of southeastern Saskatchewan. Canadian Journal of Soil Science. 80: 251-261.
Wang, C., Ross G.J., Torrance, J.K., Kodama, H. 1991. The formation of Podzolic B horizons and pedogenic imogolite as influenced by microrelief within a pedon. Geoderma. 50: 63-77.
Warren, C.J. and Dudas, M.J. 1992. Acidification adjacent to an elemental sulfur stockpile: I Mineral weathering. Canadian Journal of Soil Science. 72: 113-126.
Weil, R.R. and Brady, N.C. 2017. The nature and properties of Soils. 15th Edition. Pearson Education. 1086pp.
À Propos des Auteurs
C. James (Jim) Warren, Ph.D. P.Geo., Spécialiste des Ressources Terrestres, Ministère de l’Agriculture, de l’Alimentation et des Affaires Rurales de l’Ontario, Guelph, Ontario, Canada.

Jim est un pédologue environnementaliste avec une expérience diversifiée dans la recherche, le conseil et l’enseignement. Je suis diplômé de l’Université de Guelph (B.Sc. (Agr), Science du sol) et de l’Université de l’Alberta (M.Sc. Science du sol; Ph.D. Chimie du sol). J’ai mené des recherches en agronomie, géochimie et minéralogie à l’Université de Guelph et à l’Université de Waterloo avant de devenir consultant privé en drainage minier acide. Je suis actuellement employé en tant que spécialiste des ressources terrestres et effectue des suivis des sols et des travaux de recherche en pédologie avec le ministère de l’Agriculture, de l’Alimentation et des Affaires Rurales de l’Ontario; je suis également professeur auxiliaire à l’Université de Guelph. J’ai été chargé de cours en science du sol, minéralogie du sol, chimie du sol, géochimie et pédologie dans quatre universités (Alberta, Guelph, Waterloo et Toronto). J’ai écrit ou co-écrit plus de 100 publications dans des revues à comité de lecture; chapitres de livres, actes de conférences et rapports.
Comment me suis-je intéressé aux sols ? Eh bien, j’ai grandi dans une ferme où, adolescent, je passais beaucoup de temps sur un tracteur à faire des travaux de labour pendant lesquels j’avais beaucoup de temps pour réfléchir. Une des questions que je me suis posée pendant de longues journées sur le terrain était : « Qu’est-ce qui fait pousser les plantes ? J’ai passé les décennies suivantes à essayer de comprendre les sols pour répondre à cette question. La quête continue
. . .
Graeme Spiers, Chaire de Surveillance Environnementale et Professeur à l’École de l’Environnement, Département des Sciences de la Terre / Département de Biologie, Université Laurentienne, Sudbury, Ontario, Canada.

Graeme Spiers enseigne actuellement à l’École de l’Environnement, dans le Département des Sciences de la Terre et au Département de Biologie de l’Université Laurentienne. J’ai obtenu un B.Sc. en Sciences de la Terre et Botanique à l’Université de Waikato en Nouvelle-Zélande, et un M.Sc. et Ph.D. de l’Université de l’Alberta, avec pour spécialités la pédologie, la minéralogie de l’argile et la chimie. Du fait de mon expérience en laboratoire commercial et de consultant à travers le Canada, j’ai été associé à des programmes de recherche aux universités de l’Alberta, de Guelph, de Waterloo, des Laurentides et du Nouveau-Brunswick. Mes recherches ont donné lieu à de nombreuses publications et présentations techniques et scientifiques en chimie analytique, chimie aquatique, géologie environnementale, écologie, biologie végétale et science du sol. J’ai été éleveur laitier en Nouvelle-Zélande pendant 10 ans avant d’aller à l’université à temps plein. J’y ai obtenu mon diplôme de premier cycle sur le long terme, comme passe-temps entre les traites. À Sudbury, je participe à des travaux de recherche ayant pour but d’examiner les effets sur les sols, les rivières, les lacs et la végétation, des émissions anthropiques passées et actuelles de métaux par la fonderie de Sudbury. Je suis activement impliqué dans une variété de programmes de recherche à travers le Canada et collabore activement avec des chercheurs basés en Russie (Université d’État de Moscou et Académie des Sciences de Russie), Australie, Nouvelle-Zélande, Chine, Pérou et Afrique du Sud.
OBJECTIFS D’APPRENTISSAGE
À la fin de ce chapitre, l'étudiant devrait être en mesure:
- d'expliquer les liens qui existent entre texture du sol, densité apparente, porosité et compaction
- de décrire la façon dont les sols retiennent l’eau et les forces qui la font circuler dans le sol
- d'établir le lien entre la rétention en eau, les propriétés du sol et la disponibilité en eau pour les plantes
- de comprendre les variations de la circulation de l'eau dans le sol en fonction de son potentiel hydrique et de la distribution volumétrique des pores
- de décrire le mécanisme principal qui régit les échanges gazeux dans les sols
- d'énumérer les principaux mécanismes qui régissent le transport des solutés dans le sol de même que leur importance pour les plantes
- de décrire les propriétés thermiques du sol et les facteurs qui influencent sa température et le flux de chaleur
- d'expliquer l’effet de l’eau du sol sur sa consistance
INTRODUCTION
La physique du sol consiste à étudier ses phases solide, liquide et gazeuse de même que leurs interactions. Sa texture, sa structure et sa densité apparente reflètent la façon dont les particules minérales et organiques se combinent pour former le tout « sol », constitué d’une matrice et d’interstices non occupés par de la matière minérale solide, appelés « pores ». Ces interstices peuvent être occupés par l’air, par l’eau ou par d’autres matières gazeuses ou liquides. Les phénomènes de rétention en eau du sol et de mouvement de l’eau du sol sont déterminants dans (1) la disponibilité en eau pour les plantes et les organismes du sol, (2) l’infiltration et le drainage, (3) le ruissellement et l’érosion. Le transport des solutés — constitués de nombreux éléments nutritifs — a lieu dans la solution de sol. La circulation de l’air dans le sol et les échanges gazeux régissent les émissions de CO2 dans sol et la disponibilité de l’O2 pour les racines des plantes. Les propriétés thermiques du sol régulent la température à différentes profondeurs. Elles sont aussi en cause dans la vitesse de réchauffement du sol au printemps. La résistance du sol est influencée par sa texture et sa teneur en eau, et détermine la vulnérabilité d’un sol aux glissements de talus et à la compaction.
Les propriétés physiques du sol subissent les impacts de l’utilisation des terres à des fins d’agriculture, de foresterie ou de développement urbain. Comprendre ces impacts aide à trouver des solutions d’atténuation et de réhabilitation des sols qui concernent ces écosystèmes (agricole, forestier, urbain). La compaction du sol au cours des opérations forestières est un exemple d’impact créé sur la résistance du sol, particulièrement dans l’aire de dépôt où l’on stocke les grumes avant leur chargement par de la machinerie lourde dans des camions. Les types de sols ne présentent pas tous la même vulnérabilité à la compaction, de sorte qu’ils peuvent ne pas tous nécessiter de l’ameublissement mécanique avant la plantation d’arbres. Les aménagistes forestiers doivent tenir compte des propriétés physiques du sol, telles que sa résistance (fonction de la texture et de la teneur en eau) dans leur évaluation de ces mesures couteuses à adopter en matière de réhabilitation du sol.
Les propriétés physiques du sol subissent les impacts de l’utilisation des terres à des fins d’agriculture, de foresterie ou de développement urbain. Comprendre ces impacts aide à trouver des solutions d’atténuation et de réhabilitation des sols qui concernent ces écosystèmes (agricole, forestier, urbain). La compaction du sol au cours des opérations forestières est un exemple d’impact créé sur la résistance du sol, particulièrement dans l’aire de dépôt où l’on stocke les grumes avant leur chargement par de la machinerie lourde dans des camions. Les types de sols ne présentent pas tous la même vulnérabilité à la compaction, de sorte qu’ils peuvent ne pas tous nécessiter de l’ameublissement mécanique avant la plantation d’arbres. Les aménagistes forestiers doivent tenir compte des propriétés physiques du sol, telles que sa résistance (fonction de la texture et de la teneur en eau) dans leur évaluation de ces mesures couteuses à adopter en matière de réhabilitation du sol.
Par ailleurs, l’agriculture crée des impacts sur toutes les propriétés du sol. L’application de fumier de ferme dans les champs, par exemple, constitue une large part des 85% des émissions d’ammoniaque (NH3) qui proviennent de l’agriculture au Canada (Bittam et al., 2017). Une telle pratique découle souvent de l’excès de fumier qui se retrouve dans les champs cultivés, faute d’un meilleur usage. Cette pratique est fréquente dans les régions qui comptent beaucoup de fermes laitières et avicoles, comme la région de la vallée du Fraser en Colombie-Britannique. À des fins de diminution des impacts de l’épandage, des chercheurs d’Agriculture et Agroalimentaire Canada à Agassiz ont mis en application leur compréhension des échanges gazeux dans le système sol-plante-atmosphère. Ils ont réussi à mettre au point un épandeur de fumier qui injecte le fumier de vaches laitières dans le sol suivant une largeur entre deux rangs de maïs. L’usage de ce type de machinerie pour les épandages dans les champs cultivés aide non seulement à réduire les émissions d’oxyde nitreux (N2O) dans l’atmosphère, mais aussi à réduire les nitrates qui sont lessivés dans les eaux souterraines.
Dans les milieux urbains, la compaction du sol et le bouleversement de sa surface affectent également toutes ses propriétés. Construire une maison, par exemple, suppose (1) l’enlèvement de la végétation et la couche arable sur le chantier et (2) le compactage délibéré du sol pour réduire la sédimentation. Une fois la construction terminée, on étale dans le périmètre du chantier une fine couche de terre et l’on sème du gazon. Mais la capacité de rétention d’eau de cette mince couche de sol ne suffit pas, en été, à répondre au besoin en eau du gazon. Dans les régions les plus sèches du Canada, des villes comme Ouest Kelowna en Colombie-Britannique ont fait passer à 30 cm l’épaisseur de recouvrement obligatoire de terre arable sur un chantier de façon à augmenter la capacité de rétention d’eau pour les plantes, réduisant du coup la consommation d’eau pour les besoins extérieurs.
Ces trois exemples montrent à quel point la résolution des enjeux environnementaux rattachés au sol est impensable sans la compréhension de la physique du sol.
Le sol est un système à trois phases
Le sol intègre les trois phases de la matière : solide (matière minérale et matière organique), liquide (eau du sol, éléments nutritifs, produits chimiques) et gazeuse (p. ex. N2, O2, CO2) (Figure 4.1). L’importance relative des trois phases et leurs interactions déterminent le comportement et la fonctionnalité du sol.
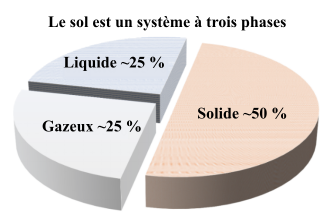
Phase solide
La phase solide du système de sol peut à la fois comprendre de la matière minérale (roches, pierres, galets, gravier, sable, limon, argile) et organique (matière organique du sol). Dans le cas idéal, la phase solide occuperait environ 50 % du volume total du système de sol (fig. 4.1). En réalité, le volume occupé par la phase solide peut toutefois varier de 30 à 60 % du volume total du sol en fonction de ses propriétés physiques et des impacts de la gestion sur lui. Par exemple, les grosses particules d’un sol sableux occupent plus de volume dans le sol que les fines particules d’un sol argileux. De même, la compaction peut faire augmenter le volume des particules solides dans un volume donné. La teneur en minéraux solides est généralement stable dans le temps, tandis que la teneur en matière organique du sol peut changer relativement rapidement. Les minéraux du sol sont constitués de particules dites primaires, telles que les minéraux cristallins (p. ex. quartz, aluminosilicates) et les gels amorphes (p. ex. oxydes, hydroxydes et oxydes hydratés de fer, d’aluminium et de manganèse). Ces particules de sol primaires interagissent. Par exemple, les gels amorphes peuvent enrober des particules cristallines pour former des particules secondaires ou des agrégats (voir chapitre 2). Les minéraux se trouvent donc constitués de particules dont la forme, la taille et la composition chimique diffèrent. Les matériaux organiques sont aussi des constituants de la phase solide du sol, mais ils occupent généralement une plus petite proportion (3 à 5 %) dans les sols minéraux. La matière organique du sol est généralement composée d’organismes vivants et de la matière résiduelle morte générée par la décomposition (à divers stades) de végétaux et d’animaux. Toute cette matière organique peut se lier avec d’autres matières organiques ou minérales pour former des unités secondaires que l’on appelle « agrégats ». Les agrégats font aussi partie de la phase solide du sol. La composition de la matière solide du sol et les caractéristiques de ses surfaces conditionnent le comportement du sol, de même que les interactions de la phase solide avec les phases liquide (eau du sol, éléments nutritifs pour les végétaux) et gazeuse (air du sol).
Phase liquide
La phase liquide du sol comprend l’eau dans laquelle se trouvent divers éléments nutritifs dissous et autres produits chimiques (les solutés du sol), le tout formant la solution de sol. La phase liquide du sol est considérée autant dans son aspect quantitatif (quantité d’eau présente dans le sol) que dans son aspect qualitatif (composition chimique de la solution de sol). Dans le cas idéal, la phase liquide occuperait 25 % du volume total du sol. La phase liquide présente un dynamisme de variation en volume que la phase solide n’a pas. Le volume de solution de sol peut varier de moins de 1 % (conditions de sécheresse totale) à près de 50 % (la solution de sol occupe tous les interstices compris entre les particules solides). Ces interstices (on dit aussi espaces ou vides) dans le sol sont connus sous le nom de « pores ». On dit qu’un sol est saturé lorsque tous ses pores sont remplis de solution de sol. Les pores non occupés par la solution de sol seront occupés par les constituants de la phase gazeuse du système (l’air du sol). On constate que les pores peuvent être occupés autant par les constituants de la phase liquide que de ceux de la phase gazeuse du sol.
Phase gazeuse
La phase gazeuse d’un sol, appelée communément « l’air du sol », est composée d’un mélange d’azote (N2), d’oxygène (O2), de vapeur d’eau et de dioxyde de carbone (CO2). Dans le cas idéal, les gaz du sol occuperaient environ 25 % du volume total du sol, mais leur nature dynamique apporte beaucoup de variations à ce volume. Les pores non occupés par la solution de sol (phase liquide) sont remplis de l’air du sol (phase gazeuse). Les gaz montrent des variations de quantité, de composition et de mobilité en fonction du temps et de leur position dans le profil du sol, et non pas en fonction de l’air atmosphérique. L’air du sol contient en général une plus grande quantité de dioxyde de carbone et une plus faible quantité d’oxygène qu’en contient l’air atmosphérique.
Fractions du sol et texture du sol
Fractions du sol
La phase (ou matière) solide du système de sol est composée de diverses particules primaires et secondaires — minérales ou organiques — dont la forme, la taille et la composition chimique ou minéralogique peuvent varier considérablement. Certaines particules sont assez grosses pour être vues à l’œil nu, tandis que d’autres ne peuvent être vues qu’au microscope. Ces dernières possèdent des propriétés colloïdales.
Les particules minérales ont été classées suivant leur grosseur (dimension ou taille). On distingue les fragments grossiers > 2 mm des fractions de terre fine (< 2 mm). On subdivise les fragments grossiers en trois classes:
- le gravier, qui comprend les particules de sol dont le diamètre varie de 2 mm à 8 cm ;
- les galets, qui comprennent les particules de sol dont le diamètre varie de 8 à 25 cm ;
- les pierres ou rochers, qui comprennent les particules de sol dont le diamètre est plus grand que 25 cm.
Si les fragments grossiers s’imposent en critère prépondérant dans l’aménagement des terres (p. ex., pour sélectionner des outils de travail du sol), ils n’ont qu’un rôle secondaire dans la capacité du sol à retenir l’eau de même qu’à stocker et à libérer les éléments nutritifs pour les plante.
On subdivise la fraction de terre fine (2 mm) en trois classes : les sables, le limon, et les argiles. Le tableau 4.1 présente les limites des classes de diamètre des particules du sol définies suivant le système canadien de classification des sols (SCCS)
Table 4.1. Désignation des fractions de sol du Système canadien de classification des sols (SCCS) définie en fonction de leur diamètre
| Fraction de sol | Diamètre des particules | |
|---|---|---|
| mm | μm | |
| Sable très grossier | 2.0-1.0 | 2000-1000 |
| Sable grossier | 1.0-0.5 | 1000-500 |
| Sable moyen | 0.5-0.25 | 500-250 |
| Sable fin | 0.25-0.10 | 250-100 |
| Sable très fin | 0.10-0.05 | 100-50 |
| Limon | 0.05-0.002 | 50-2 |
| Argile | ≤0.002 | ≤2 |
| Argile fin | ≤0.0002 | ≤0.2 |
Le terme "fraction du sol" décrit les particules minérales (sable, limon, argile) qui composent la fraction de terre fine. Le schéma de séparation des tailles de particules de sol selon le système canadien de classification des sols est présenté dans le tableau 4.1.
Sable - la plus grande division des fractions du sol, soit les particules minérales variant de 2 mm à 0.05 mm selon le SCCS. La fraction sable est subdivisée en cinq sous-fractions (très grossière, grossière, moyenne, fine et très fine). Les particules de sable sont principalement constituées de quartz minéral, mais peuvent également contenir des fragments d’autres minéraux primaires, tels que le feldspath, le mica et autres minéraux. Les particules de sable sont le plus souvent de forme sphérique, dont les bords irréguliers, durs et abrasifs, nous les font sentir granuleuses au toucher et faciles à séparer. Le sable est l’un des facteurs déterminants de la vitesse d’évacuation de l’eau dans le sol, par conséquent sur la disponibilité de l’eau pour les plantes. La part de sable d’un sol est associée à du drainage excessif et à des quantités d’eau insuffisantes pour les plantes.
Limon - fraction composée des particules de taille intermédiaire entre celles du sable et celles de l’argile. La taille des particules de limon varie de 0.05 mm à 0.002 mm (SCCS). Les particules de limon sont constituées des mêmes minéraux que ceux du sable. Elles prennent une consistance farineuse lorsqu’on les presse entre les doigts. En raison de sa plus petite taille (diamètre) que de celles du sable, la surface spécifique du limon par unité de masse est plus grande que celle du sable (notion de surface spécifique décrite plus loin).
Argile - fraction qui comprend les particules minérales de la plus petite taille (de plus petit diamètre). Aussi appelée la « fraction colloïdale ». Le diamètre des particules d’argile est inférieur à 0.002 mm et le diamètre des fines particules d’argile est inférieur à 0.0002 mm (SCCS). Les particules d’argile présentent généralement une forme qui s’apparente à une plaquette ou à une aiguille. Elles sont le plus souvent constituées de minéraux secondaires (aluminosilicates). Parmi les fines particules de terre, les particules de la taille de l’argile ont la plus grande surface spécifique par unité de masse. De plus, leur activité physico-chimique est unique. Les particules d’argile portent généralement une charge négative. Elles ont la propriété d’être souvent très réactives. Au toucher, les argiles sont très collantes, ce qui leur confère une certaine plasticité. Les particules d’argile ont la propriété d’absorber l’eau, ce qui a pour effet de faire gonfler un sol argileux, mais de le faire se rétrécir en situation de sécheresse. Tandis que des fractions de sable et de limon relativement inertes peuvent constituer le squelette du sol, les argiles sont considérées comme en étant la matière même de ce squelette.
Ensemble, ces trois fractions — sable, limon, argile — présentes suivant diverses configurations définissent ce qu’on appelle la "matrice du sol".
Texture du sol
La texture du sol définit la composition granulométrique de la matrice que l’on établit à partir de l’évaluation des proportions relatives de chacune de ses trois fractions. Cette évaluation est quantitative. La texture du sol est aussi évaluée de manière qualitative par le recours à des tests qui font appel à diverses sensations tactiles, visuelles et gustatives. Pour faciliter l’évaluation des proportions relatives de chacune des fractions de sol (sable, limon, argile) le SCCS distingue 13 classes de texture (ou classes texturales ; Figure 4.2).
La classe texturale appelée "loam" (L) est la classe qui contient des proportions à peu près égales de sable, de limon et d’argile. De manière générale, une classe texturale composée en majorité de particules d’une même taille (sable) est moins favorable à la croissance des plantes que le loam. L’information apportée par l’évaluation des classes texturales se révèle indispensable dans bien des aspects de l’utilisation des terres : détermination de la vocation des sols, gestion des sols, évaluation de leur capacité de production, etc. Nombre de propriétés du sol sont influencées par la texture : mouvement et rétention de l’eau et des solutés (éléments nutritifs) ; transfert de chaleur et aération.
L’abaque de classification texturale des sols du SCCS (Figure 4.2) permet de déterminer rapidement la classe texturale d’un échantillon de sol donné. On localise d’abord le pourcentage de sable de l’échantillon le long de l’axe inférieur de l’abaque en procédant de gauche à droite, puis on localise le pourcentage d’argile de l’échantillon le long de l’axe vertical en procédant du bas vers le haut. Depuis l’endroit correspondant à la valeur de pourcentage de sable obtenue, on projette une ligne parallèle à l’axe vertical jusqu’au point d’intersection d’une deuxième ligne projetée horizontalement à partir de la valeur de pourcentage d’argile. Le point d’intersection des deux lignes indique la classe de texture de l’échantillon de sol. Dans le cas où le point d’intersection tombe directement sur une ligne de séparation entre deux classes, on choisit habituellement la classe texturale qui contient le % d’argile le plus élevé. Le troisième axe de l’abaque présente les valeurs de pourcentage de limon. Bien souvent, il n’est pas nécessaire de connaître le pourcentage de limon puisqu’on peut déterminer la classe texturale avec seulement les pourcentages de sable et d’argile.
Surface spécifique d’un sol : définit la surface totale des particules par unité de masse de sol ou par unité de volume de particules de sol. On l’exprime généralement en mètre carré par gramme de sol (masse) ou par centimètre cube (volume) de particules de sol. La surface spécifique dépend autant de la taille des particules que de leur forme. Plus la taille des particules est petite, plus leur surface par unité de masse ou de volume est grande. Les particules de sol de forme aplatie ou allongée exposent une plus grande surface par masse ou par volume que les particules de sol de forme cubique ou sphérique. Les particules d’argile, en plus d’avoir une petite taille, revêtent la forme d’une plaquette qui leur confère une grande surface par unité de masse ou de volume de sol. Alors que les particules de sable peuvent avoir une surface spécifique d’environ 1 m2 g-1, les particules d’argile peuvent avoir une surface spécifique pouvant atteindre plusieurs centaines de mètres carrés par gramme de sol. La caractéristique fondamentale qu’est la surface spécifique de tout matériau du sol se trouve corrélée à des propriétés tout aussi importantes, telles que la capacité d’échange cationique, la rétention de l’eau, la disponibilité des éléments nutritifs et les propriétés mécaniques, y compris la plasticité, la cohésion et la résistance.
Structure du Sol
Dans le sol, les particules de matière minérale et organique sont liées par diverses forces pour former une unité structurale consistante appelée « agrégat » ou « ped ». La structure du sol renvoie à l’arrangement spatial en agrégats des particules de sable, de limon, d’argile et de particules organiques. Ces agrégats influencent la répartition des pores dans le sol. La structure du sol joue donc un rôle majeur dans le mouvement de l’eau et de l’air, dans la croissance des racines de même que dans les déplacements de la macro et méso faune.
Comment se forment les agrégats?
L’agrégation résulte de nombreux processus biologiques, physiques et chimiques qui se produisent à différentes échelles : atomique ou moléculaire, microscopique et macroscopique (échelle de perception visible par l’œil humain). La floculation, la première étape de la formation des agrégats est celle où les particules s’attirent les unes les autres grâce aux forces naturelles en présence (p. ex., liaisons électrostatiques, ponts hydroxydes OH¯, etc.). Les liaisons se produisent à l’échelle moléculaire et microscopique. La cimentation, deuxième étape de formation des agrégats permet de stabiliser les unités ainsi formées.
La figure ci-dessous illustre la formation des agrégats à différentes échelles de perception.
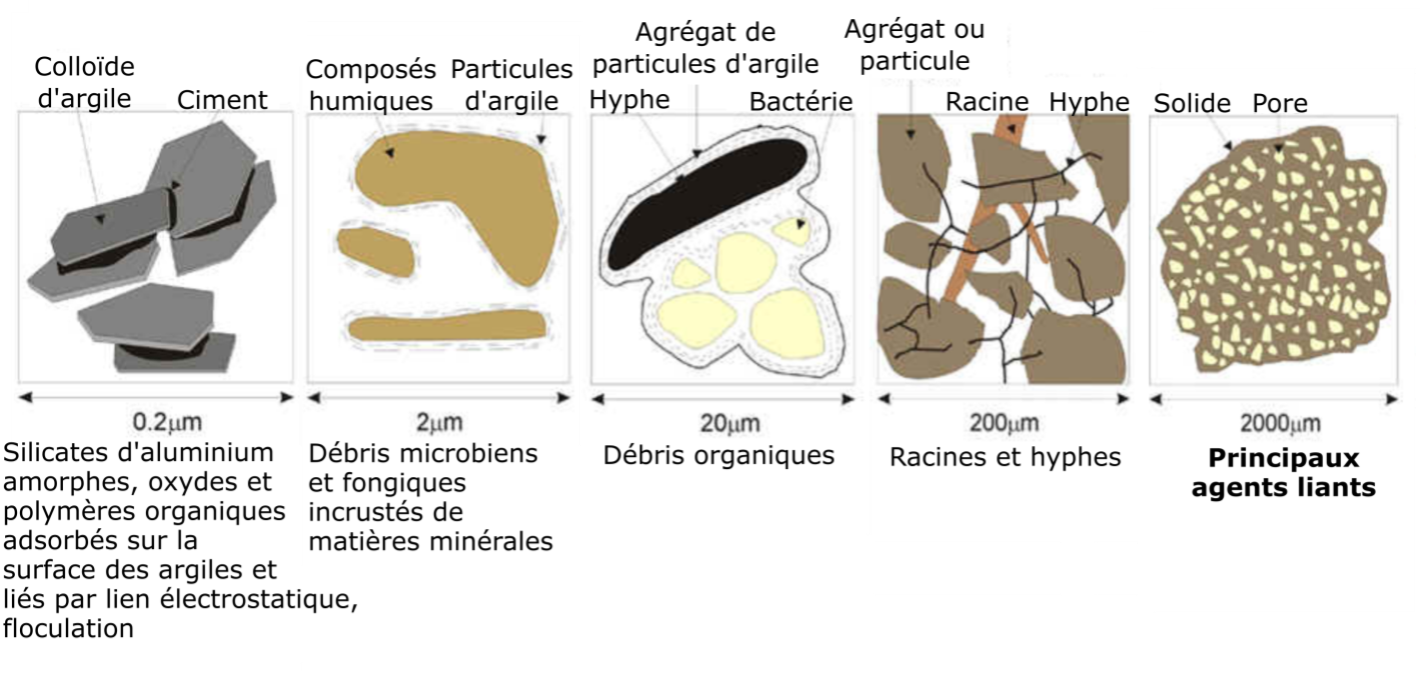
À l’échelle moléculaire, les particules d’argile (porteuses de charges négatives en surface) attirent les cations et les molécules d’eau qui ont une nature polaire. Les cations polyvalents et les molécules d’eau ont la capacité de former des liaisons électrostatiques avec les particules d’argile, tels des ponts. Lorsque le sol s’assèche, le nombre de molécules d’eau participant à la création de ces ponts diminue, de sorte que les particules d’argile se rapprochent, s’agglutinent. C’est ce qui donne lieu au phénomène de floculation.
D’autres substances participent à la création de ponts entre les particules d’argile, les composés organiques notamment. Par exemple, les molécules à longue chaîne comme les polysaccharides peuvent entrer en contact avec les particules d’argile qui les font se rassembler en une masse compacte autour d’eux. Les oxydes et hydroxydes d’aluminium et de fer peuvent jouer un rôle similaire, en créant des liens complexes entre les particules d’argile, contribuant ainsi à la formation d’agrégat.
Diverses conditions contribuent au phénomène de floculation :
- le cycle mouillage/séchage
- le gonflement/rétrécissement des particules d’argile
- le gel/dégel
Ce sont surtout dans les sols à forte teneur en argile (c.-à-d. les sols à texture plus fine) que se produira le phénomène de floculation dans ces conditions.
De leur côté, les particules de sable et de limon ne portent presque aucune charge à leur surface, mais peuvent former des microagrégats détectables à l’échelle microscopique (≤ 250 microns) en raison des particules d’argile qui les recouvrent ou des composés organiques sur lesquels elles adhèrent
Dans les sols sableux, la formation d’agrégats dépend entièrement des processus biologiques et mécaniques, qui comprennent :
- les activités d’enfouissement par les vers de terre
- la production de gels organiques par les bactéries et les champignons qui servent d’agents de cimentation et stabilisent les agrégats ; par exemple, les hyphes des champignons qui produisent une substance collante, appelée glomaline, qui agit comme agent de cimentation
- l’enchevêtrement des particules minérales (p. ex., dans un réseau de racines fines et d’hyphes fongiques
La deuxième étape, celle de la cimentation fait intervenir des processus détectables autant à l’échelle microscopique qu’à l’échelle macroscopique (≥ 250 microns), dont certains concernent également les agrégats d’argile. Ces processus favorisent la formation de gros agrégats, dits macroagrégats.
Les principaux agents de cimentation dans le sol sont :
- les composés organiques
- les revêtements d’argile
- les oxydes de Fe/Al
- les carbonates
On remarquera que ces agents jouent aussi un rôle à l’échelle moléculaire et microscopique de par leur nature moléculaire au pouvoir attractif.
On retiendra que agrégation = flocculation + cimentation, où la :
floculation définit le phénomène de rapprochement des particules de sol (surtout d’argile) par les forces naturelles. Ces forces qui comprennent les forces électrostatiques, les forces de Van der Waals, les liaisons hydrogène, les ponts hydroxydes conduisent à la formation d’agrégats microscopiques (ou amas); et où la cimentation définit l’étape de stabilisation des agrégats par un agent de cimentation, tel que les composés organiques, les carbonates, les oxydes de Fe et d’Al ou les particules d’argile.
Dispersion des argiles
La charge nette négative des particules de phyllosilicate a tendance à les faire se repousser. Lorsque ces forces répulsives sont fortes, les particules ne peuvent pas se rapprocher et on les dit dispersées. Cette répulsion mutuelle est renforcée par les cations monovalents à de faibles concentrations, car dans ces conditions, la neutralisation des charges négatives repoussantes est faible. Par conséquent, un sol dominé par les ions Na+ aura des particules d’argile à l’état dispersé, plus qu’un sol où le Ca2+ ou d’autres cations polyvalents domineront. La dispersion des particules de sol n’est pas souhaitable, car les particules dispersées conduisent à la formation de petits pores. Les infiltrations d’eau sont ainsi fortement réduites, entraînant la stagnation de l’eau à la surface du sol.
Types d’agrégats
Les sols peuvent être soit sans structure, soit n’être qu’un amas indifférencié d’agrégats. Les sols particulaires sont un exemple de sol sans structure. Ces sols sont dominés par des particules de sable et il n’y a pas de formation d’agrégats. Le manque de structure dans un sol sableux est attribuable au fait que les particules de sable, constituées de minéraux primaires tels que le quartz, portent une charge limitée, ce qui a pour effet de limiter les processus de floculation et de cimentation, particulièrement dans les sols à texture grossière.
Les sols massifs sont un autre exemple de sol sans structure. Ces sols n’ont pas d’agrégats visibles et sont typiques des argiles massives. Le manque de structure dans un sol argileux peut empêcher la croissance des plantes, car une argile massive manque suffisamment de pores moyens et gros nécessaires au développement des racines. Ce type de sol a donc un taux d’infiltration d’eau très lent.
Dans les sols structurés, les particules de sol sont constituées en agrégats ou peds distincts. Les agrégats de sol peuvent être caractérisés en termes de : forme (ou type), de taille (fine, moyenne ou grossière) et de consistance (ou de force, telle que forte, modérée ou faible). De nombreux types d’agrégats se trouvent dans les sols, dont les quatre formes principales sont sphérique, lamellaire, polyédrique (angulaire avec des bords tranchants et subangulaire avec des bords arrondis) et en forme de prisme (agrégats colonnaires orientés verticalement avec des sommets arrondis ou prismatiques à sommets plats). Ces types structurels (et certains sous-types) sont illustrés ci-dessous (Figure 4.4).

La forme des agrégats est affectée en partie par leur composition et en partie par les différences dans les processus de genèse du sol. Par exemple, les sols argileux ont souvent une structure polyédrique ; la structure colonnaire est fréquemment associée aux argiles sodiques dans les climats semi-arides ; la structure lamellaire se produit souvent près de la surface du sol en raison de l’activité de gel-dégel.
Structure du sol et espace interstitiel (ou poral)
La structure influence directement le type d’espace interstitiel dans un sol. Les pores peuvent être classés comme suit : les macropores (diamètre > 0,08 mm) se trouvent entre les agrégats ou les grains individuels dans le sol à texture grossière. Les macropores facilitent le mouvement de l’air, le drainage de l’eau et offrent de l’espace aux racines et aux organismes du sol. Les micropores (diamètre < 0,08 mm) se trouvent à l’intérieur des agrégats ou entre des particules individuelles de la taille de l’argile. Les micropores sont généralement remplis d’eau et trop petits pour permettre une bonne circulation de l’air. Le mouvement de l’eau dans les micropores est lent et une partie de l’eau qui est étroitement retenue par les colloïdes du sol n’est pas disponible pour les plantes.
Un sol argileux ayant des agrégats bien formés a tendance à avoir un équilibre entre les gros pores « d’aération et de drainage » et les petits pores de « rétention d’eau » (Figure 4.5). Une bonne structure du sol est associée à des agrégats de petite ou moyenne taille avec des pores abondants à la fois à l’intérieur des agrégats eux-mêmes qu’entre eux.
Structure du sol et mouvement de l’eau
La structure du sol influe directement sur la taille des pores ; ainsi, elle a un impact sur d’autres propriétés importantes du sol, telles que le taux d’infiltration et la rétention de l’eau, l’aération et le drainage (Figure 4.6). Un sol ayant une structure massive et des agrégats lamellaires peut inhiber l’écoulement de l’eau, tandis que l’eau se déplacera rapidement à travers les pores des sols ayant des agrégats granulaires ou particulaires.
Impact de la gestion des sols sur la structure
La structure du sol est une propriété très importante, car elle influence l’écoulement de l’eau et son aération. Pourtant, elle est facilement sujette à détérioration lorsque l’on applique des contraintes sur le sol. La contrainte peut être imposée par la culture, le piétinement par les animaux, la circulation intense attribuable à l’exploitation forestière, les motos tout-terrain, les VTT, etc. Une bonne structure (c.-à-d. un équilibre entre la présence des macropores et des micropores) est importante pour la croissance des plantes, car elle améliore le drainage et l’aération, tout en maintenant dans les agrégats de l’eau disponible pour celles-ci. Si le sol est soumis à des pratiques de gestion impliquant l’utilisation de machinerie, il peut devenir vulnérable à la compaction (réduction de l’espace poral total par broyage des agrégats et remplissage de l’espace poral). La structure peut facilement se détériorer sous les contraintes imposées par les tracteurs et les instruments et même par l’impact des gouttes de pluie ou la circulation piétonnière. La compaction est particulièrement grave lorsque les sols sont soumis à plusieurs reprises à de la machinerie lourde. Pour cette raison, les gens qui gèrent les sols pour la culture doivent respecter de nombreux principes de gestion pour maintenir une bonne structure du sol :
- Ne pas faire passer la machinerie sur le sol lorsqu’il est très humide. Lorsque la teneur en eau est très élevée, le sol ne peut pas offrir beaucoup de résistance au stress. L’équipement lourd peut facilement s’embourber dans les sols humides.
- Maintenir une teneur adéquate en calcium ou en Ca2+ en appliquant un peu de chaux (CaCO3), ce qui permet de maintenir les argiles floculées. Le calcium est un cation divalent qui crée des ponts (liaisons électrostatiques) entre les particules du sol, favorisant la formation de micro- et macro- agrégats.
- Maintenir ou ajouter de la matière organique au sol peut contribuer à améliorer la résistance des agrégats en formant des liens avec les particules du sol et à favoriser l’activité microbienne qui génère des substances qui aident à lier et stabiliser les particules.
Le travail du sol répété, en particulier dans des conditions humides, provoque la réorientation des particules et la compaction du sol juste en dessous du talon de la charrue et les roues du tracteur. La couche de sol dense qui en résulte, appelée « semelle de labour », peut entraver le drainage et l’enracinement. Un labourage en profondeur occasionnel avec une « sous-soleuse » lorsque le sol est sec peut se révéler efficace pour casser une telle semelle de labour.
Relations masse-volume
L’établissement des relations masse-volume sert à mettre en lumière de nombreuses propriétés physiques du sol, notamment sa porosité, sa densité apparente et les proportions relatives d’eau et d’air qui occupent les pores.
La masse totale de sol (fig. 4.7) comprend sa masse solide (Ms), sa masse liquide (Ml) et sa masse gazeuse (Mg). Notons cependant que la masse gazeuse est négligeable, de sorte qu’on la suppose égale à zéro.
\begin{equation}M_{t} = M_{s} + M_{l} + M_{g}\end{equation}
De même du volume total de sol $V_{t}$, qui est composé de son volume de solide ($V_{s}$), de liquide ($V_{l}$), et sa masse gazeuse ($V_{g}$). Étant donné que les volumes de liquide et de gaz constituent le volume des interstices ou pores,
\begin{equation}V_{f} = V_{l} + V_{g}\end{equation}
et, le volume total de sol,
\begin{equation}V_{t} = V_{s} + V_{l} + V_{g}\end{equation}
Dans le système SI, l’unité de masse est le kilogramme (kg) et l’unité de volume est le mètre cube (m3). De même, dans le système centimètre-gramme-seconde (CGS), l’unité de masse est le gramme (g) et l’unité de volume est le centimètre cube (cm3).
Densité ($\rho$):
La relation masse-volume d’un matériau est liée à sa densité, quelle que soit sa phase (solide, liquide ou gazeuse). On peut décrire cette relation comme suit :
\begin{equation}Masse = {Volume} \times {densité}\end{equation}
\begin{equation}Densité = \frac{Masse}{Volume}\end{equation}
L’unité de la densité est l’unité de masse sur le volume ou kg m-3 (unité SI) ou g cm-3 (unité CGS).
Densité des particules ($\rho_{p}$) : on calcule la densité des particules solides (ou du sol) comme suit :
\begin{equation}\rho_{p} = \frac{M_{s}}{V_{s}} = \frac{\text{Masse of solide}}{\text{Volume of solide}}\end{equation}
L’unité de $\rho_{p}$ est le kg m-3 (unité SI) ou g cm-3 (unité CGS). Il est plus difficile d’obtenir une valeur de $\rho_{p}$ parce que l’évaluation du volume des solides dans le sol est elle-même difficile, contrairement à l’évaluation du volume total de sol qui est beaucoup plus facile, car il comprend aussi le volume d’air et d’eau. Les valeurs de $\rho_{p}$ d’un sol minéral varieraient de 2600 kg m-3 à 2700 kg m-3 (ou de 2.6 g cm-3 à 2.7 g cm-3) si on se fonde sur les valeurs des minéraux les plus couramment trouvés dans le sol (quartz et feldspath). En règle générale, on utilise la valeur moyenne de 2650 kg m-3 (ou 2.65 g cm-3) pour calculer d’autres propriétés physiques du sol. De même, les valeurs de $\rho_{p}$ es matériaux organiques sont d’environ 1300 kg m-3 (ou 1.30 g cm-3).
La masse volumique de l’eau ($\rho_{l}$) : on calcule la masse volumique de liquide (ou de l’eau) comme suit :
\begin{equation}\rho_{l} = \frac{M_{s}}{V_{l}} = \frac{\text{Masse of liquide}}{\text{Volume of liquide}}\end{equation}
L’unité de $\rho_{l}$ est la kg m-3 (unité SI) ou g cm-3 (unité CGS). Dans la plupart des calculs, on établit la densité de l’eau à 1000 kg m-3 (ou 1.0 g cm-3).
Densité apparente sèche ($\rho_{b}$) : on calcule la densité apparente sèche comme suit :
\begin{equation}\rho_{b} = \frac{M_{s}}{V_{t}} = \frac{\text{Masse of solide}}{\text{Volume du sol total}}\end{equation}
L’unité de $\rho_{b}$ est la kg m-3 (unité SI) ou g cm-3 (unité CGS). Notons que $\rho_{b}$ ne représente que la masse des solides. Comme sa désignation l’indique, ρb comprend seulement la masse de sol sec. Les valeurs de, $\rho_{b}$ de la plupart des sols minéraux varient de 1000 à 1800 kg m-3. Cependant, les processus naturels (rétrécissement/gonflement, agrégation, gel/dégel) et les pratiques de gestion telles que le travail du sol, l’exploitation forestière ou le pâturage peuvent aussi faire varier les valeurs de $\rho_{b}$. C’est pourquoi on considère la densité apparente comme étant une propriété dynamique à la surface du sol (ou un peu en dessous de celle-ci), et plus statique plus bas dans le profil de sol. Contrairement aux sols minéraux, les sols organiques (sols avec matière organique >30% en poids) ont généralement des valeurs de $\rho_{b}$ qui varient de 800 à 1000 kg m-3, y compris des valeurs aussi faibles que 500 kg m-3 dans des sols à haut pourcentage de matière organique non décomposée (sols tourbeux).
Teneur en eau($\theta$)
La teneur en eau est la quantité d’eau (ou de liquide) présente dans un sol donné. On peut la calculer à partir de sa masse (connue sous l’appellation « teneur en eau de gravité », $\theta_{w}$) ou de son volume (connue sous l’appellation « teneur en eau volumétrique », $\theta_{v}$), en tant que rapport $M_{w}/{M_{t}$ ou $V_{w}/{V_{t}$. La valeur numérique obtenue de ce rapport est indépendante de toute unité. On la dit adimensionnelle. Cependant, il est d’usage de conserver les unités du numérateur et du dénominateur pour indiquer à partir de quel « type » de teneur en eau (gravimétrique ou volumétrique) on l’a calculée. Par exemple, l’unité de $\theta_{w}$ est en kg kg-1 ou g g-1 et l’unité de $\theta_{v}$ est en m3 m-3 ou cm3 cm-3. L’unité indique le type de teneur en eau utilisé dans le calcul. Par exemple, la valeur de 0.22 kg kg-1 du sol indique qu’il s’agit de la teneur en eau de gravité qui a été utilisée dans le calcul, soit une $\theta_{w}$ de 0.22. Sans indication d’unités ou du type de teneur en eau utilisé dans le calcul (gravimétrique ou volumétrique), la seule valeur de 0.22 pourrait mener à des erreurs d’interprétation. La valeur numérique de 0.22, en étant multipliée par 100, donne la teneur en eau en pourcentage (obtenue dans notre exemple à partir de sa masse ou teneur gravimétrique).
Teneur en eau calculée en fonction de la masse ou teneur en eau de gravité ($\theta_{w}$) : on calcule comme suit la teneur en eau de gravité en fonction de la masse du sol :
\begin{equation}\theta_{w} = \frac{M_{l}}{M_{s}} = \frac{M_{t} - M_{s}}{M_{s}} = \frac{\text{Masse de liquide (eau)}}{\text{Masse de solide}}\end{equation}
Cette valeur est adimensionnelle, mais les unités de l’équation indiquent qu’il s’agit de la teneur en eau de gravité exprimée en kg kg-1 ou g g-1.
Teneur en eau calculée en fonction du volume ou teneur en eau volumétrique ($\theta_{v}$) : on calcule comme suit la teneur en eau volumétrique :
\begin{equation}\theta_{v} = \frac{V_{l}}{V_{t}} = \frac{\text{Volume de liquide (eau)}}{\text{Volume de sol}}\end{equation}
Cette valeur est adimensionnelle, mais les unités de l’équation indiquent qu’il s’agit de la teneur en eau volumétrique exprimée en m3 m-3 ou cm3 cm-3. On notera toutefois qu’il est très difficile de mesurer le volume de liquide dans le sol. C’est pourquoi on estime le plus souvent la teneur en eau volumétrique à partir de la simple relation existant entre la teneur en eau de gravité et la densité apparente (voir eq. 14 ci-dessous).
Porosité
Dans le sol, la porosité est la fraction du sol qui n’est pas occupée par les particules solides, mais par les fractions (ou phases) liquide et gazeuse. L’appellation « volume de vide » est aussi employée. La porosité totale ($f$) donne en quelque sorte une indication du volume relatif que les pores (ou les interstices) occupent dans le sol, pores autant occupés par la fraction liquide que gazeuse du sol. Les pores occupés par la fraction de gaz portent l’appellation « porosité d’air » (ou aération) et les pores occupés par la fraction de liquide sont appelés « porosité d’eau ». La proportion de sol occupée par les pores est exprimée dans la mise en rapport du volume de porosité totale sur le volume de sol. Les unités sont exprimées en m3 m-3 (unité SI) ou cm3 cm-3 (unité CGS). On obtient la valeur du rapport en pourcentage en la multipliant par 100.
Porosité ($f$) : indique le volume total de pores (ou de vide) dans le sol ; on le calcule comme suit :
\begin{equation}f = \frac{V_{f}}{V_{t}} = \frac{V_{l} + V_{g}}{V_{t}} = \frac{\text{Volume de vide (liquide + gaz)}}{\text{Volume de sol}} = \frac{V_{t} - V_{s}}{V_{t}}\end{equation}
Les unités de $f$ en m3 m-3 (unité SI) ou en cm3 cm-3 (unité CGS). On obtient les valeurs en pourcentage en les multipliant par 100. En général, les valeurs de $f$ varient de 0.3 à 0.6 m3 m-3 dans le cas d’un sol minéral. Dans le cas des sols sableux ou de ceux dotés d’une grande proportion de particules de grande taille, les valeurs de $f$ tendent à être plus petites, tandis que les sols à dominance argileuse ou comprenant des particules plus fines présentent des valeurs de $f$ plus élevées que dans les autres sols. On gardera à l’esprit que la présence de grands pores est de loin plus corrélée avec la présence de particules de grande taille qu’avec les particules de petite taille. En revanche, les sols contenant des particules fines comptent non seulement plus de pores que les sols possédant des particules de grande taille, mais leur volume est aussi plus grand. On notera que la porosité totale ne fournit aucune information sur la distribution de la taille des pores. La partie supérieure des sols (sols superficiels ou sols de surface) a tendance à avoir une plus grande porosité que dans la partie plus profonde du profil ; le volume des pores diminue aussi, car la densité apparente du sol a tendance à augmenter avec la profondeur.
Porosité d'eau ($f_{w}$) : on calcule comme suit les pores d’eau dans le sol :
\begin{equation}f_{w} = \frac{V_{l}}{V_{t}} = \frac{\text{Volume de liquide (eau)}}{\text{Volume de sol}} = \theta_{v}\end{equation}
Les unités de $f_{w}$ sont exprimées en m3 m-3 (unité SI) ou cm3 cm-3 (unité CGS).
Porosité ou porosité d'aération ($f_{a}$) : on calcule comme suit les pores d’air dans le sol :
\begin{equation}f_{a} = \frac{V_{g}}{V_{t}} = \frac{\text{Volume de gaz (air)}}{\text{Volume de sol}}\end{equation}
Les unités de $f_{a}$ sont exprimées en m3 m-3 (unité SI) ou cm3 cm-3 (unité CGS). La porosité d’air définit la proportion de sol occupé par l’air. La porosité d’air est liée à la porosité totale et à la teneur en eau volumétrique, car l’air et l’eau partagent la même disponibilité de pores. On peut ainsi déterminer la porosité d’air en soustrayant la teneur en eau volumétrique de la porosité totale du sol. Comme la teneur en eau du sol varie en fonction du temps et de la profondeur dans le sol, il en va de même pour la porosité d’air. Les valeurs de porosité d’air seront au minimum à la suite de précipitations importantes ou de l’irrigation. En règle générale, les valeurs optimales de $f_{a}$ à la capacité au champ, après drainage, devraient être supérieures à 0.10 – 0.15 cm3 cm-3.
Relations entre les propriétés physiques du sol
S’il est possible de calculer de nombreuses grandeurs physiques à partir des relations masse-volume des trois phases du sol, il demeure parfois difficile d’évaluer la masse et les volumes des phases. Dans ces conditions, on utilise des propriétés plus faciles à mesurer pour estimer celles qui sont plus difficiles à mesurer. Les quelques équations présentées ci-dessous sont en ce sens très utiles.
On peut recourir à l’équation suivante pour exprimer la relation entre la teneur en eau volumétrique ($\theta_{v}$), la teneur en eau de gravité ($\theta_{w}$) et la densité apparente ($\rho{b}$) :
\begin{equation}\theta_{v} = \theta_{w}\rho_{b}\end{equation}
On peut recourir à l’équation suivante pour exprimer la relation entre la porosité totale ($f$), la densité apparente ($\rho_{b}$), et la densité des particules ($\rho_{p}$) :
\begin{equation}f = 1 - \frac{\rho_{b}}{\rho_{p}}\end{equation}
Enfin, on aura recours à cette équation pour exprimer la relation entre la porosité totale ($f$), la porosité d’air ($f_{a}$), et la teneur volumétrique en eau ($\theta_{v}$) :
\begin{equation}f_{a} = f - {\theta_{v}\end{equation}
Compaction
La compression d’un sol s’exerce sur ses pores qui, sous l’effet de la pression, réduisent de volume en expulsant partiellement l’air ou l’eau qu’ils renferment, d’où l’augmentation de la densité. Par exemple, sous l’action de la pression statique ou par des vibrations, les particules se trouvent réorientées à l’intérieur du même volume selon une disposition plus serrée. Un des effets de la réorientation est notamment de faire diminuer la macroporosité, ce qui réduit par le fait même la porosité totale. Dans les conditions de sol saturé, l’effet de diminution vient de l’expulsion de l’eau des macropores ; dans le cas d’un sol sec, l’effet de diminution résulte de l’expulsion de l’air. L’air s’expulse plus rapidement que l’eau, l’expulsion de l’eau résultant d’un processus en deux temps : les pores de grande taille se vident en premier suivi des autres, dans l’ordre décroissant de leur taille. Dans les conditions de sol à teneur intermédiaire en eau, l’air se trouve expulsé avant l’eau. Le terme « compaction » désigne la compression de la matrice d’un sol non saturé qui cause la réduction du volume d’air. Le terme « consolidation » désigne la compression de la matrice d’un sol saturé qui fait évacuer l’eau.
Dans le contexte de l’agriculture, un sol est considéré comme « compacté » lorsque sa porosité totale, surtout sa porosité d’air, est faible au point que l’aération du sol s’en trouve restreinte et que sa densité est forte au point de gêner 1) la pénétration des racines et 2) le drainage, en raison de la taille réduite des pores. La compaction constitue un important problème de gestion des cultures, et particulièrement le travail du sol. Le phénomène de compaction peut se produire en surface (couche durcie) ou sous la surface (horizon durci). L’usage de machinerie lourde en agriculture opérationnelle demeure une grande cause de compaction des sols attribuable à l’activité humaine. Dans les entreprises agricoles traditionnelles par exemple, jusqu’à 90 % de la superficie d’un champ peut être traversé par des roues de tracteur. Par ailleurs, l’usage de machinerie lourde en exploitation forestière est également une cause importante de compaction des sols, surtout dans les aires de dépôt (espaces dégagés en forêt où l’on débarde les arbres récoltés et où on les façonne avant leur transport hors de la forêt). Il faudra alors intervenir là où le sol compacté nécessitera une remise en état et là où la compaction aura eu pour effet de restreindre ou d’empêcher la régénération naturelle ou artificielle. La compaction des sols existe aussi en milieu urbain. Nous n’avons qu’à penser au passage répété des piétons et des bicyclettes à des endroits restreints ou aux incessantes allées et venues de la machinerie lourde en zone de construction.
Matière à réflexion !
Activité de plein air et impact sur le sol

Fouler le sol en randonnée, rouler en vélo tout-terrain, piquer sa tente dans le sol. Ces activités créent-elles un impact important sur le sol ? À la marche, le seul poids de notre corps suffit à comprimer le sol. Compression, compaction, tassement du sol… Peu importe le terme employé, l’impact créé sur le sol demeure le même : la porosité diminue et, réciproquement, la densité apparente du sol augmente. La chaîne de causes à effets enclenche les phénomènes suivants : sous la pression les macropores vont s’effondrer, empêchant ainsi l’eau de s’infiltrer, qui ne pourra plus circuler dans le sol. L’eau va plutôt ruisseler en surface pouvant aller jusqu’à provoquer le phénomène d’érosion lors d’événements de fortes pluies. Les effets de la réduction des pores priveront aussi le sol d’air. Or, un sol moins bien aéré aura des conséquences néfastes sur les microorganismes du sol et la croissance des végétaux en général. Qui plus est, la compaction créée par le piétinement à un même endroit, par exemple, le long des sentiers de randonnée et sur les sites de camping, pourra entraîner la perte de végétation. Selon des études, environ 10 m2 de superficie des sites de camping seraient caractérisés par une zone de compaction à fort impact.
Les sols des sites de camping très fréquenté subissent rapidement les conséquences néfastes de la compaction. Leur restauration complète peut prendre des décennies. On assiste toutefois à une volonté grandissante de diminuer les impacts de la compaction dans les aires de camping. Dans les campings où l’on autorise le libre emplacement des tentes, on peut exiger qu’on les déplace quotidiennement de façon à réduire l’impact potentiel de compaction sur le long terme. Des études ont montré que ces campings dits à faible impact ont réussi à réduire la compaction de la surface du sol, au point de ne plus nuire à la croissance des racines de la végétation environnante. Planter sa tente sur un sol sableux créera moins d’effet de compaction que planter sa tente sur un sol à texture plus fine. Selon les études, on conseille d’éviter de camper dans les zones plus sensibles à la compaction, de demeurer sur les sentiers de randonnée et de se rappeler que le camping écologique, c’est plus que de juste « rapporter ses déchets »!
References:
Brevik, E.C. and Tibor, M.A. 2014. Impact of camping on soil properties of Strawberry Lake, North Dakota, USA. EGU General Assembly Apr 27-May 2, 2014. Vienna, Austria.
Eagleston, H., and Marion, J.L. 2017. Sustainable campsite management in protected areas: A study of long-term ecological changes on campsites in the boundary waters canoe area wilderness, Minnesota, USA. Journal for Nature Conservation 37: 73-82. doi:10.1016/j.jnc.2017.03.004
Leave No Trace Canada. 2009. Leave no trace principles. https://www.leavenotrace.ca/principle-travel-camp-durable-surfaces
Marion, J.L. and Cole, D.N. 1996. Spatial and temporal variation in soil and vegetation impacts on campsites. Ecological Applications 6(2): 520-530.
Rétention d'eau dans les sols
Comprendre le phénomène de rétention de l’eau dans le sol et le rôle de réservoir d’eau qu’il joue pour le maintien de la vie terrestre se révèle d’une importance capitale. La capacité du sol à retenir l’eau sur une longue période permet aux plantes et aux organismes du sol à subvenir à leur besoin en eau. L’effet gravitationnel fait que l’eau du sol s’évacue de façon continue dans le sol, mais l’eau apportée par les précipitations ou l’irrigation y demeure généralement suffisamment longtemps pour que les racines des plantes aient le temps de l’absorber. Le comportement de l’eau dans le sol est tel que le déplacement des gaz qu’il induit dans les pores permet aux racines de capter l’oxygène. On distingue deux caractéristiques importantes de l’eau du sol : la quantité d’eau présente dans une quantité de sol donné (teneur en eau du sol) ; la nature des forces qui retiennent l’eau dans la matrice du sol (potentiel hydrique du sol). L’eau du sol exerce son influence sur de nombreux processus du sol : échanges gazeux avec l’atmosphère, mouvement des éléments nutritifs et des produits chimiques vers les racines des plantes, fluctuation de la température du sol, gonflement et rétrécissement. De même, les forces exercées par la matrice solide sur l’eau influenceront la façon dont l’eau (1) sera absorbée par les racines des plantes (2) sera évacuée par gravité (drainage) (3) et les solutés vaincront la gravité en adoptant une trajectoire ascendante. De tels processus sont possibles dans le sol, car il s’agit d’un milieu où l’eau se comporte très différemment de l’eau dans un verre ou dans une piscine. L’eau du sol est fortement associée aux particules solides du sol, particulièrement aux colloïdes. Les interactions produites entre les molécules d’eau et les particules du sol se trouvent à modifier le comportement des deux.
Propriétés de l'eau
L’eau influence de nombreux processus dans le sol, principalement du simple fait de sa structure moléculaire unique. L’eau est un composé moléculaire simple formé d’un atome d’oxygène lié par colavence (mise en commun d’électrons) à deux atomes d’hydrogène disposés en forme de V. Cette configuration en V non symétrique des atomes de la molécule d’eau a pour effet de produire un champ électrique. Les atomes d’hydrogène ont tendance à présenter un comportement électropositif, tandis que l’atome d’oxygène tend à présenter un comportement électronégatif, conférant à la molécule d’eau son caractère bipôle. Si la molécule d’eau présente ce caractère de polarité, c’est en raison de la répartition inégale de sa densité électronique. Une telle caractéristique dote la molécule d’eau de nombreuses propriétés qui la font jouer un rôle unique dans le sol. De par leur charge positive, les atomes d’hydrogène chargés positivement d’une molécule d’eau attirent l’atome d’oxygène chargé négativement d’une autre molécule d’eau ; de cette attirance qui se produit entre toutes les molécules d’eau résulte la formation d’une sorte d’entité groupée à l’image d’une chaîne. De plus, les molécules d’eau, de par leur polarité, se trouvent attirées par les ions (Na+, K+, Cl-) et par les colloïdes du sol (argile ou matière organique du sol).
Liaison hydrogéne
De par sa polarité, l’électronégativité de l’atome d’oxygène d’une molécule d’eau peut attirer fortement l’électropositivité de l’atome d’hydrogène d’une autre molécule d’eau à proximité. Cette force d’attraction crée un lien intermoléculaire entre les protons de l’atome d’hydrogène d’une molécule d’eau et de l’atome d’oxygène de l’autre molécule d’eau. On appelle ce lien « liaison hydrogène ». Cette force d’attraction maintient ensemble les molécules d’eau.
Forces d’adhésion et de cohésion
Les liaisons hydrogène créent les deux forces en cause dans la rétention et le mouvement de l’eau dans un sol (fig. 4.8a). On appelle « force d’adhésion » (force entre deux matières différentes) la force qui attire les molécules d’eau et les particules solides. On appelle « force de cohésion » (force entre deux matières similaires) la force qui attire les molécules d’eau entre elles. Les forces d’adhésion sont en général beaucoup plus fortes que les forces de cohésion dans le cas des molécules d’eau, car elles se trouvent fermement retenues par les solides du sol (phénomène aussi appelé « adsorption »). Ces molécules d’eau fermement adhérées aux particules solides peuvent aussi s’attacher par lien de cohésion à d’autres molécules d’eau. Ensemble, force d’adhésion et force de cohésion permettent aux solides du sol de retenir l’eau et de permettre le mouvement de l’eau dans le sol. Plus la distance augmente entre une molécule d’eau et la surface d’un solide (colloïdes du sol) plus la force d’adhésion entre les deux diminue. L’eau du sol, dans une position plus éloignée des particules de sol, peut ne pas être retenue sur leur surface.
Montée capillaire
La figure 4.8b) montre différents tubes de verre placés dans un bassin d’eau ouvert. L’eau remonte dans les tubes par capillarité, un phénomène associé à la tension de surface de tout liquide, particulièrement dans les tubes capillaires où existe l’interface gaz-liquide-solide. Comme la force d’adhésion est plus forte que la force de cohésion, l’attraction entre les molécules d’eau et la paroi de verre est plus forte que la force de cohésion entre les molécules d’eau, d’où la montée de l’eau sur la paroi de verre. C’est ce qu’on appelle le phénomène de « montée capillaire » (fig. 4.8b). La hauteur de montée capillaire de l’eau sera déterminée par le diamètre des tubes. Plus le diamètre sera petit, plus l’attraction (adhésion) entre la paroi interne (de petit périmètre) et les molécules d’eau sera forte, plus forte encore que la gravité. L’eau sera en quelque sorte maintenue par cette force d’adhésion, plus forte que la force de cohésion. À l’inverse, plus le diamètre des tubes sera grand, moins grande sera la force d’attraction (adhésion) entre la paroi interne et les molécules d’eau et, les forces de cohésion l’emportant sur les forces d’adhésion, l’eau remontera moins haut dans le tube. Par analogie, on peut voir les pores dans le sol comme autant de tubes assemblés en faisceaux. Les particules de petite taille (argile) créeront des faisceaux de plus petits pores que les particules de grande taille (sable), d’où la montée de l’eau plus importante dans les pores des premiers.
Teneur en eau du sol
La teneur en eau du sol est la quantité d’eau présente dans le sol. La quantité maximale d’eau qu’un sol peut contenir est égale au volume total des pores. Lorsque tous les pores sont remplis d’eau, on dit que le sol est saturé d’eau. On désigne alors la teneur en eau de celui-ci comme étant la teneur maximale en eau (fig. 4.9). Lorsqu’un sol saturé d’eau entre dans un processus d’assèchement, divers facteurs contribuent à le freiner, tels que la force d’attraction (force d’adhésion) qui agit entre les molécules d’eau et la surface des particules de sol. Dans les conditions de saturation, on observe deux phénomènes : les pores sont tous remplis d’eau et des couches de molécules d’eau superposent la surface des particules du sol. La force d’adhésion qui agit entre les différentes couches de molécules d’eau et les particules du sol n’est pas la même pour toutes les couches. Les couches de molécules d’eau les plus proches de la surface des particules de sol seront plus fortement attirées que les couches les plus éloignées. La force d’adhésion diminue donc avec la distance : plus une couche de molécules d’eau est loin de la surface des particules, moins forte est la force d’adhésion entre les deux. Dans un sol caractérisé par des pores de grande taille, les molécules d’eau qui se trouvent les plus éloignées de la surface des particules de sol sont faiblement retenues par elles ; la force d’adhésion est donc faible. Dans des conditions où un sol saturé peut se drainer librement, la force gravitationnelle dépasse la force d’adhésion entre les particules de sol et les molécules d’eau. Le sol se draine jusqu’à ce que la force gravitationnelle et la force d’adhésion entre les particules de sol et les molécules d’eau s’égalisent. La quantité d’eau qui peut être drainée par la force gravitationnelle est appelée « eau de gravité ou eau gravitationnelle » (fig. 4.9). L’eau de gravité sort rapidement d’un sol, si tant est que les plantes ou les microbes ne puissent en faire usage.
Une fois l’eau de gravité drainée, l’eau encore présente dans le sol demeure retenue à l’intérieur des pores de petite taille (micropores ou capillaires). En raison de leur petite taille, les micropores ont la propriété de retenir l’eau par la force d’adhésion qui s’exerce entre les particules de sol et les molécules d’eau et par la force de cohésion qui s’exerce entre les molécules d’eau elles-mêmes. L’eau des micropores peut être extraite par des forces externes, telles que celles que génèrent les racines des plantes. Mais pour que les plantes puissent extraire des micropores les molécules d’eau les plus rapprochées de la surface des particules solides du sol, il faut que ses forces soient plus fortes que la force d’adhésion. Or, la pression (ou force) que peuvent exercer les racines sur les particules pour en extraire l’eau est limitée. La quantité d’eau que les plantes peuvent extraire du sol est appelée eau disponible pour les plantes ou réserve d’eau utile du sol (RU). L’eau maintenue par les forces de cohésion dans les micropores (ou capillaires) est dite « eau capillaire » (fig. 4.9). Les molécules d’eau qui demeurent fixées aux particules du sol principalement par la force d’adhésion forment l’eau hygroscopique. L’eau hygroscopique ne représente qu’une faible quantité d’eau dans le sol. De plus, elle demeure non disponible pour les plantes, car ces dernières ne disposent pas de la force nécessaire pour vaincre la force d’adhésion qui maintient les molécules d’eau fixées sur la surface des pores. Il est possible de mesurer cette force d’adhésion qui agit entre la surface du sol et les molécules d’eau ; il s’agit de la mesure du potentiel capillaire du sol, décrite plus loin dans le chapitre. On emploie aussi les expressions « potentiel de matrice du sol » ou « potentiel matriciel du sol » pour parler de cette force.
Dans le processus d’assèchement d’un sol saturé, l’eau de gravité s’écoule en premier, suivie de l’eau disponible pour les plantes, laquelle diminue à mesure que l’assèchement se poursuit jusqu’à ce qu’elle ne soit plus disponible pour les plantes ; seule l’eau hygroscopique demeure encore dans le sol. L’étape du processus d’assèchement qui marque la fin de l’évacuation de l’eau de gravité et le début de l’évacuation de l’eau disponible pour les plantes est appelée capacité au champ (fig. 4.9). La capacité au champ désigne la teneur en eau que le sol contient encore deux à trois jours après avoir été saturé (par irrigation ou par les précipitations), une fois que l’eau de gravité a été évacuée. C’est l’eau disponible pour les plantes. L’étape du processus d’assèchement qui caractérise le moment où les plantes ne sont plus en mesure d’extraire l’eau du sol est appelée point de flétrissement permanent. L’eau n’est plus disponible pour les plantes. Passé le point de flétrissement permanent, les plantes ne pourront plus récupérer, même si on ajoutait de l’eau dans le sol.
Potentiel hydrique du sol
La mesure de l’humidité du sol se révèle nécessaire pour qui a besoin d’établir un bilan hydrique du sol, gérer l’irrigation, évaluer la sécheresse et prévoir les crues. Dans la section précédente, la teneur en eau du sol a été définie comme étant la quantité (en masse ou en volume) d’eau présente dans une masse ou un volume donné de sol, respectivement la teneur en eau de gravité et la teneur en eau volumétrique. L’eau du sol est dotée de sa propre énergie, ce qui la fait participer à son propre mouvement dans le sol. Comme toute masse, l’eau n’échappe pas aux lois de la physique, y compris celle qui fait se déplacer une masse d’un endroit de haut potentiel à un endroit à faible potentiel.
Qu'est-ce que l'énergie?
Toute masse a de l’énergie. Les unités d’énergie sont les joules (J). Contrairement à la masse, cependant, l’énergie est difficile à définir et à conceptualiser. Voici une définition de l’énergie prise dans un manuel d’introduction à la physique :
« [l’énergie est une] quantité associée à un état (ou à une condition) d’un ou de plusieurs objets »
« [l’énergie est] un nombre que nous associons à un système d’un ou de plusieurs objets. Si une force fait bouger un objet, le nombre change aussi »
Ces définitions semblent un peu trop générales, du moins pas aussi précises que peut l’être la définition de la masse (la quantité de matière d’un objet). Le concept d’énergie devient plus clair lorsqu’on l’associe : (1) au mouvement : énergie cinétique ; (2) à la séparation d’un objet d’un autre objet à l’énergie connue : énergie potentielle (gravitationnelle, électrique) ; à la (3) température : énergie thermique.
Potentiel hydrique du sol - unités
Étant donné la lenteur du mouvement de l’eau dans le sol (vitesse très faible), l’énergie cinétique de l’eau du sol est moins importante que son énergie potentielle. C’est pourquoi on ne considère que l’énergie potentielle de l’eau que l’on appellera simplement « potentiel hydrique du sol », soit celle induite par la gravité ou l’électricité, tel qu’il est mentionné ci-dessus. À l’instar de l’évaluation de la masse d’eau, qui est effectuée par unité de masse de sol (teneur en eau de gravité) ou par unité de volume de sol (teneur en eau volumétrique), l’évaluation du potentiel hydrique du sol l’est également par unité d’eau du sol :
- énergie potentielle par unité de masse : J kg-1
- énergie potentielle par unité de volume : J m-3 or Pa
- énergie potentielle par unité de poids : J N-1 or m (equivalent height of water)
Peu importe l’unité que l’on choisira dans l’expression du potentiel hydrique du sol, les trois unités se convertissent l’une à l’autre.
L’évaluation de l’énergie potentielle se fait toujours par rapport à un bassin de référence
On calcule le potentiel hydrique du sol en établissant la différence entre l’énergie potentielle de l’eau du sol et celle de l’eau à la surface d’un bassin de référence. Le bassin de référence utilisé dans le calcul du potentiel hydrique du sol correspond à l’eau pure à la surface d’un bassin, à la pression atmosphérique et à une élévation donnée.
Un exemple d’eau à la surface d’un bassin de référence : celle d’un bécher rempli d’eau posé sur une table de travail dans un laboratoire. Par convention, on attribue la valeur zéro au potentiel d’eau du bassin de référence pour qu’il soit toujours le même, quelles que soient ses unités (c.-à-d. J kg-1, le Pa, ou le m). De plus, la valeur zéro permet d’évaluer rapidement si le potentiel hydrique du sol est supérieur, égal ou inférieur à celui du bassin de référence.
Définition officielle du potentiel hydrique du sol et notion de travail
La Soil Science Society of America a défini officiellement le potentiel hydrique du sol comme suit :
« Quantité de travail à effectuer pour transporter une quantité infinitésimale [par quantité unitaire en masse, en volume ou en poids] d’eau pure d’un bassin de référence donné, d’une élévation donnée et à la pression atmosphérique, jusqu’à l’eau d’un sol situé à une distance verticale donnée par rapport au bassin [sans changement de sa température] » (Soil Science Society of America, 1997).
En d’autres termes, le potentiel hydrique du sol correspond à la quantité de travail qu’il faut exercer sur l’eau d’un bassin de référence jusqu’à ce qu’elle atteigne un endroit déterminé dans le sol, sans changement de sa température.
En savoir plus sur la notion de travail aidera à comprendre ce qu’on entend au juste par « potentiel hydrique du sol ». En physique, on définit le travail comme étant le produit d’une force (action) appliquée sur un objet, suffisante pour faire changer son énergie potentielle. Par exemple, si on soulève une boîte depuis un plancher jusque sur une table, le potentiel de gravité de la boîte a augmenté proportionnellement à la distance d’élévation entre le plancher et la table.
On peut se demander la somme de travail (d’effort) qu’il faudrait fournir pour transporter l’eau depuis un bassin de référence jusqu’à un endroit déterminé dans le sol.
Recourons à un phénomène familier pour répondre à cette question. Prenons les éponges. Comme le sol, elles ont des pores et la capacité de retenir l’eau. Imaginez que vous prenez une éponge sèche pour essuyer un peu d’eau qui a été déversée sur une table. Par analogie, identifiez l’éponge au sol et identifiez l’eau sur la table à l’eau dans le bassin de référence. Sans aucun effort de votre part, l’eau s’est comme spontanément infiltrée dans l’éponge. Quel effort (ou somme de travail) avez-vous dû déployer pour que l’eau s’infiltre dans l’éponge ? Aucun. Ce « aucun » effort dans ce contexte est appelé « travail négatif » sur l’eau, l’éponge l’ayant tout fait à votre place. Imaginez maintenant l’effort que vous auriez à fournir pour faire l’inverse du « travail » de l’éponge, c’est-à-dire faire sortir l’eau de l’éponge. Vous presseriez l’éponge. Soyez honnête : auriez-vous réussi à faire sortir toute l’eau de l’éponge, jusqu’à sa dernière molécule ? Non, quelle que soit la pression appliquée sur l’éponge, cette dernière retiendrait toujours une petite quantité d’eau. L’eau que vous avez réussi à sortir de l’éponge a exigé un certain effort à faire de votre part. Dans ce contexte, on dit que vous avec fait un « travail positif » sur l’eau pour qu’elle sorte de l’éponge. Mais comme l’eau n’est pas complètement sortie de l’éponge, vous auriez eu à « travailler » un peu plus pour arriver à l’extraire jusqu’à sa dernière molécule.
Poursuivant avec l’analogie de l’assimilation de l’éponge à un sol et de l’eau sur la table à un bassin de référence, on peut conclure que le potentiel de l’eau dans le sol est inférieur au bassin de référence. Pourquoi ? Parce qu’un travail négatif est en cause dans l’absorption de l’eau et un travail positif est en cause dans l’extraction. On peut aussi en conclure que c’est la force d’attraction (d’adhésion) entre les particules du sol et les molécules d’eau qui influent sur le potentiel hydrique du sol. Pour exprimer quantitativement le potentiel total de l’eau du sol, il faut considérer toutes les forces qui agissent sur l’eau du sol et qui influencent par le fait même son potentiel hydrique. On les présente au tableau 4.2.
Table 4.2. Forces composantes du potentiel hydrique total du sol
| Composantes de forces | Appellation de la composante de force du potentiel hydrique du sol | Bassin de référence | Importance du potentiel de la composante dans le bassin de référence |
|---|---|---|---|
| Force d’adsorption entre les particules du sol et l’eau, force capillaire (au-dessus de la nappe phréatique ; sol non saturé) | Potentiel matriciel, hm |
Eau à pression atmosphérique | 0 |
| Force osmotique des solides dissous | Potentiel osmotique, ho |
Eau pure1 | 0 |
| Force de gravité, élévation | Potentiel d’eau libre ou de gravité, hg |
Eau à une élévation de référence | 0 |
| Pression de l’eau sus-jacente (sous la nappe phréatique ; sol saturé) | Potentiel de pression, hp |
Eau à pression atmosphérique | 0 |
| Tout ce qui précède | Potentiel total, H = hm + ho + hg + hp |
Eau pure à pression atmosphérique à une élévation de référence | 0 |
| 1Dans l’eau pure, le nombre de particules de soluté dissoutes est égal à zéro, le potentiel osmotique y est donc nul. | |||
Le potentiel matriciel résulte de l’attraction des molécules d’eau vers les solides du sol, attribuable aux phénomènes d’adsorption et de capillarité (similaire à l’éponge). Comme le potentiel matriciel réduit la liberté du mouvement de l’eau par rapport au bassin de référence, il s’agit d’un potentiel négatif. Un sol sec a un potentiel matriciel très faible (c.-à-d. très négatif), tandis qu’un sol humide a un potentiel matriciel plus élevé (c.-à-d. plus près de zéro).
Le potentiel osmotique résulte de l’attraction de l’eau vers les solutés, ces derniers ayant pour effet de réduire le niveau d’énergie de l’eau dans la solution du sol. Le potentiel osmotique peut avoir un impact sur l’absorption de l’eau par les racines des plantes dans les sols salins, mais la concentration de solutés dans la plupart des sols est faible, de sorte qu’elle n’influence pas le mouvement de l’eau.
Le potentiel gravitationnel est l’attraction, par simple gravité, de l’eau vers le plus bas du profil de sol. On le définit par rapport à une élévation donnée (p. ex., le haut du profil du sol). Si la nappe phréatique a remonté jusqu’à la surface, le sol se trouve alors complètement saturé en eau. Dans cette condition, le potentiel matriciel est inexistant, mais il existe un potentiel de pression qui résulte du poids de l’eau sus-jacente (comme la pression exercée sur les tympans lorsqu’on nage jusqu’au fond d’une piscine).
À n’importe quel endroit donné dans un profil de sol, le potentiel hydrique du sol total résulte de la somme de chacune de ses composantes. L’eau du sol chemine toujours d’un endroit à fort potentiel total vers un endroit à faible potentiel total. Cependant, ce ne sont pas toutes les composantes du potentiel hydrique du sol qui contribuent au potentiel hydrique total du sol en toutes circonstances. Par exemple, le potentiel de pression est nul au-dessus de la nappe phréatique, parce que la force qui augmente le potentiel de pression est la force de la pression de l’eau présente sous la nappe phréatique. Mais, sous la nappe phréatique, le potentiel matriciel est nul, parce que les forces qui agissent sur le potentiel matriciel, l’adsorption et la capillarité, sont très faibles dans les sols saturés.
Courbe de rétention en eau dans le sol
L’eau du sol est à la fois masse et énergie. La teneur en eau du sol (en masse ou en volume) est évaluée à partir de la teneur en eau gravimétrique (eau de gravité) ou volumétrique, et le potentiel hydrique du sol est évalué par rapport au potentiel connu d’un bassin de référence. Quelle relation existe-t-il entre la teneur en eau dans le sol (en masse ou en volume) et le potentiel hydrique du sol?
Déjà à la fin des années 1800, Edgar Buckingham se posait la question. Il a découvert qu’il existait une relation entre la teneur en eau du sol et le potentiel hydrique du sol. Il a montré qu’une relation positive existait entre le potentiel matriciel de l’eau du sol et la teneur en eau du sol.
Cet Américain spécialiste en sciences du sol avait compris qu’une masse se déplaçait d’endroits à fort potentiel total vers des endroits à faible potentiel total. Il en a ainsi déduit que si le potentiel total de l’eau du sol était le même à tous les endroits, il n’y aurait aucun déplacement d’eau. Buckingham voulait montrer que le potentiel total en eau du sol était le même à chaque endroit. Pour ce faire, il créa lui-même un dispositif de mouvement d’eau du sol. Il disposa des colonnes de sol d’une hauteur de 1.2 m, chacune dans un bassin d’eau peu profonde. Au début, l’eau dans les bassins s’est mise à remonter dans les colonnes remplies de sol sec, puis, après une période assez longue, l’eau ne s’est plus déplacée. À ce moment, le potentiel total de l’eau était partout égal dans les colonnes de sol.
Buckingham a supposé que c’était les forces capillaires et d’adsorption (les forces matricielles) qui faisaient remonter l’eau des bassins dans les colonnes de sol et qu’elle pouvait y demeurer malgré l’effet de la gravité. Cette supposition l’a mené à conclure que le potentiel gravitationnel et le potentiel matriciel étaient les composantes clés dans l’évaluation du potentiel hydrique total du sol. En fait, dans le cas d’un potentiel total égal à tous les endroits, le potentiel gravitationnel et le potentiel matriciel doivent être égaux, mais être de signes opposés. La hauteur à laquelle l’eau monte dans les colonnes de sol dépend de la taille des pores (fig. 4.6a). L’élévation de l’eau sera plus élevée dans une colonne contenant un sol à petits pores parce leur force capillaire est plus grande que celle d’un sol doté de pores de plus grande taille. En mesurant la teneur en eau du sol à différents endroits le long des colonnes de sol, Buckingham a pu montrer la relation entre le potentiel matriciel (adsorption et capillarité) et la teneur en eau du sol. C’est cette relation, entre la teneur en eau et le potentiel matriciel, qu’exprime la courbe de rétention d’eau du sol (fig. 4.10b).
Chaque sol présente une courbe de rétention d’eau unique, qui peut être exprimée mathématiquement suivant la fonction : $h_{m}(\theta)$ ou $\theta(h_{m})$ (c.-à-d. que le potentiel matriciel est fonction de la teneur en eau et vice versa). Pour mieux comprendre pourquoi chaque sol est caractérisé par une courbe de rétention d’eau unique, les physiciens du sol ont adopté le modèle de tube capillaire des pores du sol. Ce modèle suppose que les pores du sol se comportent de la même manière que les tubes capillaires, même si les pores du sol n’ont pas la même forme qu’eux. Comme l’a observé Buckingham, si l’on place une colonne de sol sec dans un bassin d’eau, l’eau s’élèvera spontanément dans la colonne de sol, tout comme l’eau montera dans un tube capillaire placé dans un bassin d’eau. Ce sont les forces d’adsorption et de capillarité (les forces associées au potentiel matriciel) qui font monter l’eau dans la colonne de sol. Par conséquent, on peut relier le potentiel matriciel au rayon efficace des pores remplis d’eau en modifiant comme suit l’équation de la hauteur de montée capillaire :
\begin{equation}h_{m} \leq \frac{-0.15}{R\ast}\end{equation}
où, $h_{m}$ est le potentiel matriciel (cm H2O), et $R\ast$ (cm) est le rayon efficace des pores d’eau.
Par conséquent, la courbe caractéristique de l’eau du sol constitue une représentation de la répartition volumétrique cumulative des pores du sol.
Disponibilité en eau pour les plantes
L’importance d’acquérir des connaissances sur la disponibilité en eau pour les plantes et les microorganismes passe nécessairement par l’acquisition des connaissances sur la façon dont les sols la retiennent et la libèrent. Un tel bagage d’information scientifique de base se révèle indispensable aux pratiques de gestion de l’eau en agriculture, en agroforesterie, à la remise en état des terres et des écosystèmes urbains.
La notion de réserve d’eau utile du sol (RU) peut servir à évaluer la quantité d’eau du sol disponible au besoin des plantes. Rappelons que la RU se définit comme résultant de la différence de teneur en eau volumétrique entre une limite supérieure, appelée capacité au champ, et une limite inférieure, appelée point de flétrissement permanent (fig. 4.9). L’eau disponible est celle à laquelle les plantes ont accès, soit celle présente dans leur zone d’enracinement. L’évaluation de la disponibilité en eau se fait dans cette zone (en m d’eau par profondeur d’enracinement).
Une telle information sert à déterminer la quantité d’eau nécessaire pour que l’on puisse refaire au moment opportun (par irrigation en mm) la réserve utile en eau. Il faut supposer dans l’évaluation que l’eau est également disponible pour les plantes pour la durée comprise entre la capacité au champ et le point de flétrissement permanent.
Irriguer, c’est viser à assurer la disponibilité en eau pour les plantes par application artificielle d’eau sur les terres de façon à corriger tout déficit pluviométrique qui leur créerait du stress. Il n’y aurait que les deux tiers de la RU qui seraient effectivement disponibles pour les plantes avant qu’elles ne commencent à souffrir de stress hydrique, car il peut survenir un moment où les plantes peuvent extraire plus rapidement l’eau du sol que ce dernier est en mesure de les approvisionner (Reicosky et Ritchie, 1976 ; Rekika et al., 2014). Pour répondre au besoin en eau des plantes, il faut donc autant leur assurer un apport continu en eau, que du moment opportun où l’eau deviendra disponible aux racines après avoir cheminé dans la matrice du sol. Il est désormais possible de relever le défi que pose la gestion de l’irrigation grâce au recours des tensiomètres et des sondes d’humidité, qui permettent d’évaluer la disponibilité en eau dans la matrice du sol. Dans le passé, on a beaucoup utilisé l’irrigation par aspersion et par rigoles pour refaire les réserves en eau du sol. De jours, on utilise de plus en plus l’irrigation goutte à goutte et l’irrigation souterraine, ce qui permet de gérer la disponibilité en eau des plantes dans un volume de sol limité. En adaptant l’irrigation aux conditions de croissance de chaque culture, nous optimisons à la fois la productivité des cultures et l’utilisation de l’eau (Caron et coll., 2015, 2017).
MOUVEMENT DE L'EAU DU SOL
Débit d’eau dans un sol saturé et dans un sol non saturé
L’eau chemine dans le sol depuis des endroits à fort potentiel total vers des endroits à faible potentiel total. Les deux équations qui décrivent l’intensité du débit vertical de l’eau dans le sol dans des conditions de sol saturé et non saturé sont très similaires. L’écoulement de l’eau dans les sols saturés (ceux dont les pores sont tous remplis d’eau) est décrit par la loi de Darcy :
\begin{equation}q_{w} = -K_{s}\frac{\Delta H}{\Delta z}\end{equation}
où $q_{w}$ est l’intensité du débit d’eau dans le sol en unités de $\frac{m^3 H_2 O}{m^2 soil\cdot s}$; $K_{s}$ iest appelée « conductivité hydraulique à saturation » et ses unités sont les $\frac{m^3 H_2 O}{m^2 soil\cdot s}$ par unité de gradient hydraulique, et $\frac{\Delta H}{\Delta z}$ est le gradient hydraulique (résultant de la somme des changements de pression et du potentiel gravitationnel sur un certain intervalle de profondeur $\Delta z$); le gradient est adimensionnel si $h_{p}$ est exprimé en m H2O.
Le signe négatif dans la loi de Darcy indique la direction du débit vers le bas (descendant). L’écoulement ascendant (de direction z croissante) est de signe positif et l’écoulement descendant (de direction z décroissante) est de signe négatif. L’écoulement (ou flux) descendant est associé à des gradients positifs ($\frac{\Delta H}{\Delta z}$ > 0), ce qui signifie que le potentiel total diminue avec la profondeur.
Edgar Buckingham a observé que la plupart du temps les sols étaient dans des conditions de saturation non complète (seule une fraction des pores était remplie d’eau). Dans ces conditions de saturation partielle, le gradient hydraulique variait en fonction des changements qui pouvaient survenir dans le potentiel matriciel du sol et que la conductivité hydraulique variait en fonction de l’humidité dans le sol. Le résultat suit la loi du débit de Buckingham-Darcy :
\begin{equation}q_{w} = -K(\theta)\frac{\Delta H}{\Delta z}\end{equation}
où, $K(\theta)$ est la conductivité hydraulique déterminée en fonction de la teneur en eau, que l’on appelle simplement « conductivité hydraulique non saturée », et $\frac{\Delta H}{\Delta z}$ est le gradient hydraulique (résultat de la somme des changements de pression et du potentiel gravitationnel sur un certain intervalle de profondeur, $\Delta z$)
Conductivité Hydraulique
La conductivité hydraulique (K) est une propriété hydraulique du sol qui joue en faveur de l’écoulement de l’eau dans le sol. Chaque sol est doté d’une conductivité hydraulique unique qui est liée à la distribution de la taille de ses pores. La figure 4.11 montre K en fonction du potentiel matriciel de trois échantillons de sol de texture différente. Dans des conditions de saturation, les sols qui possèdent de grands pores continus auront une conductivité hydraulique beaucoup plus élevée que ceux caractérisés par de petits pores discontinus. À mesure que la teneur en eau dans un sol diminue, la conductivité hydraulique diminue aussi parce que : (1) le volume total des pores d’eau diminue ; et (2) la distribution de la taille des pores d’eau diminue (fig. 4.12). Par conséquent, la conductivité hydraulique du sol est fonction de la teneur en eau volumétrique.
À saturation, la conductivité hydraulique de chaque sol atteint la valeur maximale (Ks), soit la conductivité hydraulique à saturation, mais elle diminue de façon exponentielle à mesure que le sol s’assèche (l’axe des y, celui de la conductivité hydraulique, est sur une échelle logarithmique). Les sols à texture grossière sont dotés de pores de plus grande taille que les sols à texture plus fine, telle que le loam et le loam limono-argileux. C’est pourquoi les valeurs de K des premiers sont plus élevées.
Processus hydrologiques et bilan hydrique du sol
Infiltration et ruissellement
L’infiltration décrit le phénomène du cheminement de l’eau (des précipitations ou de l’irrigation) dans le sol suivant ses diverses voies naturelles d’emprunt (fig. 4.13). On peut quantifier le phénomène par la détermination du taux d’infiltration. On obtient le taux d’infiltration en évaluant le volume d’eau qui entre dans une section transversale de sol par unité de temps (mm h-1). Le taux d’infiltration est fonction du temps que met la matrice du sol à se saturer. La capacité d’infiltration de l’eau dans le sol dépendra à la fois de la distribution de la taille des pores, de la présence ou non de continuité entre eux, de même que de la teneur en eau avant infiltration. Au moment où le débit d’eau dépasse la capacité d’infiltration du sol, du ruissellement se produit. Le ruissellement constitue un processus important d’érosion hydrique, car les eaux de ruissellement transportent et redistribuent les sédiments le long des pentes en plus de créer des rigoles ou des ravins à la surface du sol.
Drainage, percolation en profondeur, montée capillaire
Dans le bas d’un profil de sol, le mouvement de l’eau vers le haut ou vers le bas découle de l’interaction entre trois composantes : la percolation en profondeur, le drainage et la montée capillaire. La percolation en profondeur définit le mouvement descendant de l’eau dans le profil de sol. Le drainage : on en distingue deux catégories : (1) le drainage de surface (ou superficiel), qui consiste à faire évacuer l’excès d’eau à la surface du sol, soit par canaux naturels améliorés, fossés creusés ou par modelage du terrain et (2) le drainage souterrain, qui consiste à intercepter l’eau qui percole dans le sol au moyen de canalisations enfouies dans le sol, généralement constituées de tuyaux en plastique ondulé. La montée capillaire : sous l’action des fluctuations de la nappe phréatique, l’eau souterraine peut remonter vers le haut du profil de sol par saturation ou par capillarité. Ces trois agents en cause (percolation en profondeur, drainage, montée capillaire) dans les mouvements ascendant et descendant de l’eau dans le profil de sol, sont fonction du temps. On les exprime en unités de volume d’eau par unités de surface par unités de temps.
Bilan de l’eau dans le sol
Le bilan hydrique du sol représente l’équilibre entre les intrants et les extrants d’une unité de sol donnée (Éq. 20). Le stockage de l’eau du sol (ΔS)est l’une des composantes d’un bilan hydrique. Cette composante correspond à la mesure du changement de volume d’eau dans une unité de sol donnée sur une période de temps ; on l’exprime généralement en hauteur équivalente d’eau par unité de temps (mm day-1).
\begin{equation}P + I - ET + Q_i - Q_o - \Delta S - DP = 0\end{equation}
où, $P$ = précipitations; $I$ = irrigation; $ET$ = évapotranspiration; $DP$ = drainage et percolation profonde; $Q_i$ = autre débit entrant (en provenance de la montée capillaire et des fluctuations de la nappe phréatique); et $Q_o$ = autre débit sortant (ruissellement, écoulement latéral).
Procéder à des mesures pour l’établissement d’un bilan hydrique du sol (à l’aide d’appareils de mesure appelés lysimètre) prend beaucoup de temps et requiert beaucoup de ressources. On peut palier cet inconvénient en estimant le bilan hydrique du sol à partir des mesures de base qui caractérisent un site donné, telles que la teneur en eau du sol et son potentiel hydrique, certaines propriétés du sol, de même qu’à partir de données météorologiques sur un intervalle de temps donné.
TRANSPORT DE SOLUTÉ
Les engrais, les pesticides et les stimulateurs de croissance sont fréquemment utilisés en agriculture. Ces produits agrochimiques ont largement contribué à l’augmentation des rendements des cultures depuis les années 1950 (Ritchie et Roser 2013). Les engrais et les pesticides sont aussi couramment utilisés en milieu urbain (p. ex. pelouses privées, parcs urbains), ainsi que sur les terrains de golf et les terrains de jeux. Cependant, l’application excessive de produits chimiques associée à une gestion inadéquate a pour effet de réduire l’efficacité de l’utilisation des éléments nutritifs par les plantes, d’augmenter les émissions de gaz à effet de serre et de polluer l’environnement. Pour qui entend optimiser l’application et la gestion de ces produits, être au fait du déplacement des produits chimiques dans le sol jusqu’aux racines des plantes est une connaissance essentielle.
Comme il est décrit dans la section précédente, le sol intègre les trois phases de la matière (solide, liquide et gazeuse). Le transport des produits chimiques dissous (solutés présents dans la solution de sol) a lieu dans la phase liquide du sol, ce qui permet aux racines des plantes de pouvoir les absorber. On distingue deux principaux mécanismes de transport du soluté dans le sol : par diffusion moléculaire et par débit massique.
Diffusion moléculaire
Toute molécule présente dans un liquide possède une énergie cinétique interne qui la maintient dans un mouvement continu, même si le liquide est immobile. Les trajectoires empruntées par cette molécule résultent autant de son mouvement aléatoire propre que des collisions avec les molécules voisines dotées de cette même énergie cinétique interne. Ce mouvement global d’agitation est connu sous le nom de mouvement brownien. Si le mouvement d’une molécule individuelle peut prendre toutes les directions, le mouvement collectif des molécules, lui, tend à préférer une seule direction, soit celle qui fait mouvoir les molécules et ions en zones de concentration élevée vers les zones de concentration faible (fig. 4.14). De plus, plus la différence de concentration des molécules et ions est élevée entre les deux zones, plus le déplacement (vitesse de diffusion) des molécules et ions entre chaque zone est rapide. On mesure quantitativement la vitesse de diffusion, (le flux), comme étant la masse de soluté qui traverse perpendiculairement une unité de surface par unité de temps (kg m-2 s-1), suivant la loi de diffusion :
\begin{equation}q_s = -D_s\frac{\Delta C}{\Delta x}\end{equation}
oú: $q_s$ est le flux chimique (kg m-2 s-1); $\Delta C$ et $\Delta x$ expriment la différence de la distance ($\Delta x = x_2 - x_1$) entre deux zones de concentration (masse de soluté)entre deux zones de concentration (masse de soluté) ($\Delta C = C_2 - C_1$) separated by distance $\Delta x$ ($\Delta x = x_2 - x_1$); et $D_s$ est la constante de proportionnalité appelée « coefficient de diffusion » (m2 s-1).
Les ions et molécules (le soluté) finissent par se répartir uniformément dans tout le milieu, lequel finit par avoir une concentration homogène. Dans le cas de l’application d’une bande d’engrais dans le sol, l’engrais finirait par se répartir uniformément, en raison du phénomène de diffusion, suivant un gradient de concentration qui le ferait se déplacer de la zone de concentration élevée en solutés (là où se trouve la bande d’engrais) vers les zones environnantes de concentration moins élevée. La figure 4.15 montre la progression de la diffusion de l’engrais depuis le moment de l’application en bande (représentée par la bande verticale marron - concentration maximale d’engrais). La diffusion progresse de façon uniforme de part et d’autre de la bande, telle que l’illustre l’aplatissement horizontal uniforme de la courbe de distribution de l’engrais en fonction de la distance de la bande. L’aplatissement correspond à la réduction des valeurs de concentration avec le temps.
Le coefficient de diffusion détermine non seulement le gradient de concentration, mais aussi la vitesse de diffusion et le flux chimique. Le volume des pores d’eau, la tortuosité des pores et la force d’adhésion ont tous un effet sur le coefficient de diffusion des molécules dans le sol.
Débit massique
Le soluté se déplace par diffusion dans la solution de sol, même lorsque cette dernière est immobile. Toutefois, toute forme de pression appliquée sur l’eau dans le sol la fera se déplacer suivant un gradient de potentiel élevé à moins élevé, en entraînant avec elle tous les constituants en solution ou en suspension. Ce déplacement (ou transport) de l’eau et des solutés (la solution de sol) qui cause un changement de son potentiel est appelé débit massique. La quantité de soluté transportée par unité de surface de sol suivant le débit massique est donnée par le flux de soluté ($q_s$, kg m-2 s-1), qui résulte de la multiplication du débit d’eau par la concentration de soluté dans la solution :
\begin{equation}q_s = q_wC\end{equation}
où, $q_w$ est le débit d’eau (m s-1) et $C$ la concentration de soluté dans l’eau (kg m-3).
Transport des éléments nutritifs jusqu’aux racines
Les plantes sont dotées d’un réseau très développé de racines fines qui leur permettent d’absorber l’eau et les éléments nutritifs. Par simple contact direct avec le sol, les racines sont capables d’intercepter les éléments nutritifs. Cependant, les racines fines des plantes ne sont généralement en contact direct avec le sol que de l’ordre d’environ 1 % du volume de ce dernier. C’est pourquoi les racines ont besoin de mécanismes supplémentaires pour accéder aux éléments nutritifs et à l’eau des 99 % restants du volume du sol.
Débit massique - L’eau de l’évapotranspiration des plantes provient de l’eau du sol que leurs racines fines sont en mesure d’absorber. Les poils racinaires des racines fines ont par ailleurs la capacité d’absorber — en les attirant vers eux — les éléments nutritifs dissous que l’eau (ou solution de sol) transporte sous forme de solutés. Le débit massique est le phénomène qui permet aux plantes d’absorber les éléments nutritifs mobiles dans la solution de sol, tels que l’azote et le soufre. Les plantes augmentent leur probabilité d’absorber les éléments nutritifs dissous dans la solution de sol si leur concentration augmente près de leur zone racinaire. L’application d’engrais sert ce but.
Diffusion - Les racines utilisent en premier lieu les éléments nutritifs qui se trouvent sur le pourtour direct de leurs poils racinaires. À mesure que la concentration des éléments nutritifs diminue autour des poils racinaires, celle juste un peu plus loin des racines fines peut cependant être encore élevée. Le gradient de concentration créé entre les deux zones déclenchera alors le phénomène de diffusion : les éléments nutritifs de la zone la plus concentrée (juste un peu plus loin des racines fines) migreront vers la zone la moins concentrée (au pourtour des poils racinaires). Le flux chimique est proportionnel au gradient de concentration, de sorte que si le gradient de concentration entre la zone racinaire et le sol environnant est élevé, le flux des éléments nutritifs en direction de cette zone sera rapide. Il est possible de créer un gradient de concentration important si les éléments nutritifs se trouvent aussi près que possible de la zone racinaire et aussi souvent que faire se peut.
Dans des conditions où le taux d’évapotranspiration des plantes est élevé, le débit massique devient le mécanisme dominant de transport des éléments nutritifs jusqu’à la zone d’absorption par les racines. En revanche, dans des conditions d’évapotranspiration faible des plantes, la diffusion prime comme mécanisme de transport des éléments nutritifs jusqu’à la zone d’absorption par les racines. Le phénomène de diffusion peut être entravé dans la situation où les poils racinaires, faute d’une densité adéquate, ne suffiraient pas à absorber tous les éléments nutritifs disponibles.
AÉRATION DU SOL ET ÉCHANGE GAZEUX
L’aération du sol est déterminante dans la croissance des plantes et des microorganismes. Stockage d’air, échange gazeux et absorption de gaz sont tous des processus qui ont lieu dans les pores du sol. Le processus de stockage de l’air a principalement lieu dans les grands pores. Le processus d’échange gazeux se produit selon la capacité de diffusion des gaz dans les pores, laquelle capacité dépend de leur forme, leur taille et la connectivité entre eux. Le stockage ou l’épuisement d’un gaz particulier varie en fonction de son gradient de concentration et de l’activité biologique du milieu, autrement dit de la présence de puits et de sources d’origine microbienne ou végétale. L’oxygène et le dioxyde de carbone (e.g., O2, CO2) sont deux exemples de gaz dont les processus d’échange ont lieu dans les pores d’air étant donné leur faible solubilité dans l’eau.
L’air contenu dans les pores du sol se trouvera chassé par compression des phases solide et liquide. Cette action de densification fait diminuer le volume total des pores occupés par l’air en plus de redistribuer les pores restants d’une tout autre façon. La densité apparente s’en trouve alors augmentée. Deux raisons expliquent pourquoi le volume des pores occupés par l’air dans un sol est une donnée utile. La première est qu’il est facile de mesurer une telle donnée et d’en interpréter le résultat ; la seconde vient du fait que ce sont dans les pores occupés par l’air qu’ont lieu les principaux échanges gazeux dans le sol. La mesure du volume de pores occupés par l’air sert notamment à caractériser la productivité potentielle des horizons de surface d’un sol.
Composition de l’air du sol
La composition en gaz dans le sol varie en fonction de l’activité biologique (sources et puits) qui a lieu dans le sol et de la facilité avec laquelle les gaz du sol peuvent se mélanger avec les gaz de l’air atmosphérique (N2, O2, CO2). Près de sa surface, le sol échange facilement des gaz avec ceux de l’atmosphère. Dans le sol, les racines et les microorganismes utilisent l’O2 et le remplacent par duet le remplacent par du CO2. Il s’ensuit que l’air du sol devient plus faible en O2 et plus élevé en CO2. Quand l’eau augmente dans un sol, l’air diminue, et par voie de conséquence, les échanges gazeux qui ont lieu entre le sol et les plantes diminuent. La teneur en humidité peut augmenter jusqu’à créer un milieu asphyxiant pour les plantes, ce qui, entre autres, nuit à leur croissance. Ces conditions dites anoxiques ralentissent aussi toute l’activité biologique.
Diffusion des gaz
Les gaz dans le sol ne sont pas immobiles. Les principaux agents de déplacement des gaz dans le sol comptent l’humidité, les changements de température, la pression barométrique et la diffusion. La diffusion est de loin le principal agent de déplacement des gaz dans le sol. Une molécule de gaz donné dans l’air du sol (O2 or CO2) se déplacera selon un gradient de concentration donné selon la première loi de diffusion de Fick.
Première loi de Fick :
\begin{equation}J = -D(\frac{dC}{dx})\end{equation}
où, $J$ = flux de diffusion (g m-2 s-1); $D$ = capacité de diffusion du gaz dans le sol (m2 s-1); et $\frac{dC}{dx}$ = gradient de concentration du gaz (g m-2).
Par exemple, le flux de diffusion (J) d’O2 découlera du gradient de concentration ($\frac{dC}{dx}$), donné par le déplacement de l’oxygène d’une zone de pression partielle supérieure (ou concentration) dans l’atmosphère juste au-dessus du sol à une zone de pression partielle inférieure (concentration) dans le sol, résultant de la respiration racinaire et microbienne (fig. 4.16). Le flux de diffusion est déterminé par la capacité de diffusion ($D$) en fonction d’un gradient de concentration donné. La capacité de diffusion de l’oxygène dans l’air est d’environ 10 000 fois plus élevée que celle de l’oxygène dans l’eau. Dans le sol, la capacité de diffusion de l’oxygène est fonction du volume des pores occupés par l’air, de leur taille et de la connectivité entre eux.
Plus profond dans le sol, la concentration d’un gaz (O2 or CO2) dépend à la fois de sa capacité de diffusion et de l’activité biologique, notamment la respiration. Les figures 4.16 et 4.17 montrent le changement graduel par diffusion des concentrations d’oxygène O2 et de dioxyde de carbone (CO2) attribuable à l’activité racinaire ou microbienne. La diminution d’O2 et à une capacité de diffusion plus faible (il y a moins d’échange d’O2) et à une capacité de diffusion plus faible (il y a moins d’échange d’O2 avec l’air de l’atmosphère). Les pratiques de gestion des cultures, telles que l’irrigation, l’ajout de matière organique de même que le passage répété de machinerie lourde à un même endroit sont tous des agents perturbateurs de l’activité biologique et de la capacité de diffusion des gaz dans le sol. À mesure que la teneur en eau du sol augmente, la capacité de diffusion du gaz diminuera et, par conséquent, le pourcentage d’O2 sera plus faible en profondeur (c’est-à-dire en passant des lignes vertes aux lignes bleues sur la figure 4.17). L’ajout de compost augmentera l’activité biologique, augmentera ainsi la consommation d’O2 et se traduira par un pourcentage plus faible d’oxygène en profondeur (c’est-à-dire en passant des lignes pointillées aux lignes continues sur la figure 4.17). Le compactage du sol réduira la porosité à l’air, diminuera D et entraînera une diminution du pourcentage d’oxygène en fonction de la profondeur. D’autres échanges gazeux ont lieu à travers le continuum sol-plante-atmosphère selon des processus de diffusion et de transformation biologique semblables à ceux de l'O2, du méthane (CH4), de l'oxyde nitreux (N2O), et de l'ammoniac (NH3) (trois gaz qui jouent tous un rôle important dans le changement climatique).
TEMPÉRATURE DU SOL ET FLUX DE CHALEUR
La température conditionne beaucoup les processus biologiques et chimiques qui ont lieu dans le sol, tels que l’activité microbienne, le cycle des éléments nutritifs, la croissance des racines et les émissions de gaz à effet de serre. La germination des graines et la croissance des plantes sont également conditionnées par la température. De plus, le gel et le dégel des quelques centimètres de la surface du sol, phénomène commun dans les régions froides, peut être préjudiciable à certaines plantes. La température idéale de germination de la plupart des graines de cultures est d’environ 10°C. Toutefois, les légumineuses, certains pois, lentilles et pois chiches peuvent germer à des températures avoisinant 5°C. Où, la germination hâtive, en étant associée à une saison de croissance plus longue, peut signifier une augmentation de rendement. En revanche, la germination tardive peut signifier une récolte tardive. Une récolte tardive sera davantage exposée au risque de gel et aux températures froides, ce qui aura pour effet de réduire considérablement le rendement d’une culture donnée. Par conséquent, la température du sol est une caractéristique fondamentale autant pour la compréhension des processus du sol que pour sa saine gestion.
La manière dont se comporte le sol sous l’influence des variations de température dépend de ses propriétés thermiques. Ainsi chaque sol a son propre rythme de réchauffement au printemps, de refroidissement à l’automne, au cours de l’hiver, rythme qui est plus accéléré dans les horizons de surface que dans les horizons sous-jacents. On distingue la capacité calorifique du sol de sa conductivité thermique ; les deux varient en fonction de la teneur en minéraux par rapport à la teneur en matière organique contenue dans le sol et en fonction de sa teneur en eau.
Capacité calorifique du sol : capacité du sol à retenir ou à emmagasiner la chaleur. Il s’agit de la quantité de chaleur (énergie) qu’il faut fournir pour élever d’un degré la température d’un volume unitaire de sol. Cela signifie que plus la valeur de capacité calorifique est élevée, plus d’énergie sera nécessaire pour induire un minimum de changement de température. Il faut beaucoup d’énergie pour élever la température de l’eau et moins d’énergie pour élever la température de l’air. On dira que la capacité calorifique de l’eau est élevée, et celle de l’air, négligeable. La capacité calorifique dans le sol augmente de façon linéaire avec l’augmentation de la teneur en eau (fig. 4.18b). Par conséquent, un sol sec se réchauffera plus rapidement qu’un sol humide, car l’énergie nécessaire pour élever la température d’un sol sec est inférieure à celle nécessaire pour élever un sol humide. La capacité calorifique de la matière organique solide dépasse celle des particules minérales solides. De plus, étant donné que les couches organiques sont beaucoup plus poreuses que les couches minérales d’un sol, la capacité de stockage de chaleur des sols organiques demeure plus faible que celle des sols minéraux. Les sols à haute capacité calorifique résistent mieux aux changements de température.
Conductivité thermique (λ) : désigne la capacité d’un sol à transmettre la chaleur lorsqu’il est soumis à un gradient de température. La conductivité thermique des particules minérales dans un sol est de loin supérieure à celle de la matière organique ; et la conductivité thermique de l’eau est de loin supérieure à celle de l’air (tableau 4.3). Les couches organiques, comme on en observe dans les sols forestiers, ont tendance à avoir un λ inférieur à celui des sols minéraux, parce que non seulement ces couches renferment beaucoup de pores, mais ils sont en très grande majorité occupés par de l’air. Dans un sol minéral, la conductivité thermique est influencée par sa teneur en eau. Comme on peut l’observer à la figure 4.18a, la conductivité thermique augmente rapidement dans la couche minérale avec l’augmentation de la teneur en eau. En effet, en adhérant aux particules solides, l’eau pelliculaire favorise le transfert de chaleur d’une particule solide à l’autre, l’eau étant de loin un meilleur conducteur de chaleur que l’air. Ainsi, les sols humides transféreront mieux la chaleur que les sols secs, alors que les sols secs — parce qu’ils sont faiblement conducteurs de chaleur — font de bons isolants.
Table 4.3. Conductivité thermique (λ) des composants du sol (adapté de de Vries, 1963)
| Composante du sol | λ (J/msKo) |
|---|---|
| Quartz | 8.368 |
| Minéraux argileux | 2.930 |
| Eau | 0.594 |
| Matière organique | 0.251 |
| Air | 0.026 |
Influence de la conductivité thermique et de la capacité calorifique sur la température du sol
La température traduit l’agitation des molécules (leur énergie cinétique) d’un corps lorsqu’on lui fournit de la chaleur. Le soleil est la principale — pour ne pas dire l’unique — source de chaleur du sol. En réchauffant l’eau dans le sol et en stimulant les propriétés thermiques de ce dernier, le soleil agit jusque dans ses profondeurs. Comparons les régimes de température de surface de deux sols minéraux caractérisés par la même texture, la même teneur en matière organique, la même porosité et densité apparente, mais de teneur en eau différente (un sol humide par rapport à un sol sec). Les deux sols ont la même température. Le soleil brille par une chaude journée d’été. En atteignant la surface des deux sols, le rayonnement solaire se transforme en énergie thermique. La température augmentera à la surface des deux sols. Le sol humide transférera la chaleur plus rapidement vers le bas du profil de sol que le sol sec, étant donné la conductivité thermique plus élevée du premier. De plus, la surface du sol humide nécessiterait une augmentation plus forte de chaleur que dans le cas de la surface du sol sec pour faire augmenter sa température de 1°C. En revanche, la plus faible conductivité thermique du sol sec l’empêchera de transférer la chaleur vers le bas du profil de sol de sorte que la chaleur (dit aussi contenu calorifique) s’accumulera à sa surface. Dans ces conditions, même une petite augmentation de contenu calorifique entraînerait une forte augmentation de la température à la surface du sol sec. On en déduit que la température de la surface du sol sec fluctuera de façon plus considérable que la température de la surface d’un sol humide.
CONSISTANCE ET RÉSISTANCE DU SOL
La consistance définit la résistance des matériaux constitutifs d’un sol à la déformation qui résulte des forces physiques de cohésion (qui collent ensemble les particules d’un même matériau) et d’adhésion (qui collent ensemble des particules de matériaux différents). La consistance d’un sol est principalement donnée par sa teneur en argile et sa teneur en eau (fig. 4.19). Imaginons, en s’en rapportant à la figure 4.19, que nous prenons de l’argile sèche entre nos doigts et que nous y ajoutions graduellement de l’eau tout en travaillant l’échantillon. Dans un premier temps, l’échantillon s’apparente à une poudre libre qu’aucune pression des doigts ne parviendra à donner consistance. Puis, de petites portions de matière s’agglutineront à mesure que l’on y incorporera un peu plus d’eau. Ce changement de consistance marquera la limite dite de retrait. L’ajout graduel transformera l’échantillon en une matière plus malléable, plus cohésive, autrement dit de moins en moins friable ; on pourra presque former un moule qui ne se fragmentera pas sous la pression des doigts. Ce changement de consistance correspondra à la limite de plasticité de l’échantillon. Mais l’ajout d’encore un peu d’eau transformera progressivement l’échantillon en une masse plus compacte, encore plus facile à mouler, qui conservera sa forme sous la pression des doigts ; la limite de plasticité sera alors atteinte, car on saura qu’en ajoutant encore de l’eau, l’échantillon ne serait plus autant « plastique ». Et effectivement, l’incorporation additionnelle d’eau à l’échantillon aura pour effet de faire disparaître l’état plastique du sol ; on ne pourra plus le modeler à souhait. Il aura même tendance à « couler » entre les doigts quand on le pressera. C’est comme si la consistance de l’échantillon de sol se trouvait entre un état plastique et liquide ; la limite de liquidité est atteinte.
Ces limites qui marquent un changement de consistance dans un échantillon de sol (limite de retrait, limite de plasticité et limite de liquidité) sont appelées limites d'Atterberg limits (fig. 4.19). L’écart qui sépare la limite de plasticité et la limite de liquidité d’un sol est une donnée qui se révèle utile dans le domaine de l’ingénierie. En effet, contrairement aux spécialistes du sol qui cherchent à se documenter sur la résistance des sols à la déformation dans le but de les préserver à des fins de cultures diverses, les ingénieurs cherchent plutôt à se documenter sur la résistance du sol à un stress à des fins de construction.
SOMMAIRE
- Le sol est appréhendé comme un système qui intègre les phases solide, liquide et gazeuse de la matière en constante interaction. L’importance en volume de chaque phase et les interactions qui se produisent entre elles déterminent le comportement et les mécanismes du sol.
- La composition de la matière minérale (% de sable, de limon et d’argile) et la matière organique déterminent la porosité du sol, laquelle lui confère ses capacités de drainage, de rétention d’eau et d’aération.
- Les forces d’adhésion et de cohésion régissent la rétention de l’eau dans un sol.
- La notion de potentiel hydrique du sol sert à déterminer le mouvement de l’eau dans le sol.
- La notion de conductivité hydraulique définit la facilité avec laquelle l’eau s’écoule dans le sol.
- Les notions de diffusion et de débit massique décrivent les deux principaux mécanismes de transport des solutés dans un sol.
- La diffusion est le principal mécanisme de transport des gaz dans un sol.
- Les notions de conductivité thermique et de capacité calorique servent à évaluer la vitesse à laquelle un sol se réchauffe ou se refroidit.
EXERCICES PRATIQUES
- Donnez la définition des termes ci-dessous :
- Surface spécifique
- fraction de terre finE
- potentiel hydrique du sol
- Définissez chacun des termes ci-dessous et relevez les différences entre les paires de termes :
- texture du sol et structure du sol
- densité des particules et densité apparente
- capacité au champ et point de flétrissement permanent
- Tracez la courbe caractéristique de rétention d’eau a) d’un sol sableux b) d’un sol loameux (inclure les étiquettes des axes).
- Donnez la définition du potentiel hydrique du sol et expliquez le phénomène de déplacement de l’eau en fonction d’un gradient de potentiel hydrique donné.
- Énumérez les forces constitutives du potentiel hydrique total du sol, puis donnez leur définition.
- Quelle est la relation entre le potentiel matriciel et la taille des pores du sol ?
- Comment peut-on obtenir de l’information sur la taille et la distribution volumétrique des pores d’un sol à partir de sa courbe caractéristique de rétention d’eau ?
- Décrivez brièvement chacun des deux modes de transport des solutés dans un sol : par débit massique et par diffusion.
- Expliquez le phénomène de diffusion de l’oxygène dans le sol en fonction de l’occupation relative des pores, par l’air ou par l’eau.
- Comment le phénomène de compaction influence-t-il la distribution d’oxygène dans le sol ?
- Dans les régions tempérées (comme à Vancouver en C.-B.), où les hivers sont doux et humides, les sols peuvent prendre beaucoup de temps à réchauffer au printemps. Pourquoi les sols humides réchauffent-ils moins rapidement que les sols plus secs ?
EXEMPLES TRAVAILLÉS
Exemple d’étude de sol n° 1 - Densité apparente et teneur en eau de gravité
Au cours de l’été, Zineb, étudiante à l’Université de la Colombie-Britannique, a prélevé une carotte de sol à la ferme de l’UBC. L’échantillon a été pesé, séché 24 heures au four à 105ºC (ou jusqu’à masse constante), puis pesé de nouveau.
 Photo de Zineb Bazza, University of British Columbia, Vancouver @ CC BY |
Elle a obtenu les données suivantes : | |
| Volume de l’échantillon (carotte de sondage) | 330 cm3 | |
| Poids de la sonde pédologique en métal | 56.0 g | |
| Poids de l’échantillon de sol frais + sonde | 499.4 g | |
| Poids sec de l’échantillon de sol | 386.1 g | |
Calculez, à l’aide des équations présentées dans le chapitre, la densité apparente et la teneur en eau de gravité de cet échantillon de sol.
Étape 1 : Calcul de la densité apparente
On sait que $\rho_b = \dfrac{M_s}{V_t}$
où $M_s$ = mass of solides et $V_t$ = volume total de sol
Ainsi, $\rho_b = \dfrac{M_s}{V_t} = \dfrac{386.1\,g}{330\,cm^3} = 1.17\,g\,cm^{-3}$
Étape 2 : Calcul de la teneur en eau de gravité
On sait que $\theta_w = \dfrac{M_l}{M_s} = \dfrac{M_{sol\;frais} - M_{sol\;séché}}{M_s} $
où
Masse de sol frais = (masse sol frais + masse sonde pédologique) - masse sonde pédologique = $499.4\,g -56.0\,g = 443.4\;g$
Ainsi, $\theta_w = \dfrac{443.4\,g - 386.1\,g}{386.1\,g} = \dfrac{57.3\,g}{386.1\,g} = 0.15\,g\,g^{-1}$
Exemple d’étude de sol n° 2 - Teneur en eau de gravité et teneur en eau volumétrique
Un échantillon de sol prélevé dans la région de Chaudière-Appalaches au Québec a une teneur en eau de gravité (θw) de 0.19 g g-1 et une densité apparente (ρb) de 1.26 g cm-3. Quelle est la teneur en eau volumétrique (θv) de cet échantillon en cm3 cm-3 ? Utilisez les équations présentées dans le chapitre.
On sait que $\theta_v = \dfrac{V_l}{V_t}$ et $\theta_w = \dfrac{M_l}{M_s} = \dfrac{M_t - M_s}{M_s}$
Et, $\rho_l = \dfrac{M_l}{V_l}$ ou $V_l = \dfrac{M_l}{\rho_l}$ et $\rho_b = \dfrac{M_s}{V_t}$ or $V_t = \dfrac{M_s}{\rho_b}$
Ainsi, $\theta_v = \dfrac{V_l}{V_t} = \dfrac{\frac{M_l}{\rho_l}}{\frac{M_s}{\rho_b}} = \dfrac{M_l \cdot \rho_b}{M_s \cdot \rho_l} = \theta_w\dfrac{\rho_b}{\rho_l}$
Et maintenant, si on considère la densité du liquide (l’eau dans ce cas-ci) = 1 g cm-3 (en unité CGS), alors on peut simplement écrire $\theta_v = \theta_w\rho_b$
Dans le cas de l’échantillon de sol prélevé au Québec, on aurait :
$\theta_v = \theta_w\rho_b = 0.19 g g^{-1} \times \dfrac{1.26 \frac{g}{cm^3}}{1 \frac{g}{cm^3}} = 0.24 \frac{cm^3}{cm^3}$
Exemple d’étude de sol n° 3 - Densité apparente et porosité
Un sol loameux du sud de l’Alberta a une porosité de 48 %. Quelle est sa densité apparente en kg m-3 ? Effectuez votre calcul à l’aide des équations présentées dans le chapitre.
Si on suppose une densité de particules de $2.65\frac{kg}{m^3}$ et une porosité de 48 % ou $0.48\frac{cm^3}{cm^3}$, la densité apparente est de $1,378\frac{kg}{m^3}$
On doit réorganiser l’équation pour calculer la porosité :
Porosité $= 1 - \frac{\rho_b}{\rho_s}$ où $\rho_b$ est la densité apparente du sol et $\rho_s$ est la densité des particules du sol.
Par conséquent : $\rho_b = 2.65 \frac{g}{cm^3} - (2.65 \frac{g}{cm^3} \times 0.48) = 1.378 \frac{g}{cm^3} = 1,378 \frac{kg}{m^3}$
Exemple d’étude de sol n° 4 - Mouvement de l’eau dans le sol
Miles donne le cours d’initiation aux sciences du sol à l’Université de l’Alberta. À des fins pédagogiques, il a effectué des mesures de la tension d’un sol (T) à 3 profondeurs, avant et après un grand événement de pluie. Malheureusement, il a perdu certaines mesures.
A) Remplissez les deux tableaux ci-dessous en recourant à cette information additionnelle :
- la solution du sol ne contient pas de sels (ions) et le sol est bien aéré
- les trois endroits où les mesures ont été prises dans le profil du sol se trouvent au-dessus de la nappe phréatique
- la surface du sol constitue le point (ou l’élévation) de référence
[indice : rappelez-vous que $T = - \text{potentiel matriciel}$ ($h_m$)]
Tableau
| Point | Profondeur dans le sol (m) |
T (m) | hg (m) | hm (m) | ho (m) | hp (m) | ht (m) | Sens de l’écoulement (  ) ) |
|---|---|---|---|---|---|---|---|---|
| A | 0 | 2.3 | ||||||
| B | -0.1 | 2.45 | ||||||
| C | -0.25 | 2.9 |
Tableau II
| Point | Profondeur dans le sol (m) |
T (m) | hg (m) | hm (m) | ho (m) | hp (m) | ht (m) | Sens de l’écoulement ( ) |
|---|---|---|---|---|---|---|---|---|
| A | 0 | 5.2 | ||||||
| B | -0.1 | 4.4 | ||||||
| C | -0.25 | 4.65 |
B) Déterminez, à partir des calculs que vous avez effectués, à quel événement de pluie (avant ou après) correspond chacun des tableaux. Expliquez brièvement.
Solution:
- détermination du potentiel de gravité $h_g$ – étant donné que le point (ou l’élévation) de référence est situé à la surface du sol, et que les unités sont en m d’eau, la valeur du potentiel de gravité $h_g$ correspond à la même valeur que celle de la profondeur du sol ; elle devient de plus en plus négative à mesure que l’on descend dans le profil du sol (autrement dit que l’on s’éloigne du point référence)
- rappelons que le potentiel matriciel ($h_m$) = $-\; T$
- étant donné que la solution de sol ne contient aucun sel (ion), le potentiel osmotiqu $h_o = 0$
- étant donné que le sol est bien aéré et que les points de mesures (endroits des mesures) sont situés au-dessus de la nappe phréatique, $h_p = 0$
- rappelons que $h_t = \Sigma(h_g + h_m + h_o + h_p)$
- appelons que l’eau du sol se déplace d’un endroit de haut potentiel vers un endroit de faible potentiel ; il est ainsi possible de déterminer le sens de l’écoulement de l’eau
Tableau I
| Point | Profondeur dans le sol (m) |
T (m) | hg (m) | hm (m) | ho (m) | hp (m) | ht (m) | Sens de l’écoulement ( ) |
|---|---|---|---|---|---|---|---|---|
| A | 0 | 2.3 | 0 | -2.3 | 0 | 0 | -2.3 | |
 |
||||||||
| B | -0.1 | 2.45 | -0.1 | -2.55 | 0 | 0 | -2.55 | |
|
||||||||
| C | -0.25 | 2.9 | -0.25 | -3.15 | 0 | 0 | -3.15 |
Tableau II
| Point | Profondeur dans le sol (m) |
T (m) | hg (m) | hm (m) | ho (m) | hp (m) | ht (m) | Sens de l’écoulement (  ) ) |
|---|---|---|---|---|---|---|---|---|
| A | 0 | 2.3 | 0 | -5.2 | 0 | 0 | -5.2 | |
 |
||||||||
| B | -0.1 | 2.45 | -0.1 | -4.4 | 0 | 0 | -4.5 | |
|
||||||||
| C | -0.25 | 2.9 | -0.25 | -4.65 | 0 | 0 | -4.9 |
B) Le tableau I contient les données d’après l’événement de pluie, parce que la direction de l’écoulement de l’eau va en descendant dans le profil de sol (c.-à-d. du point A vers le point C). Le mouvement descendant de l’eau signifie que l’eau pénètre dans le sol.
Le tableau II contient les données d’avant l’événement de pluie. La flèche indique que l’eau se déplace vers le haut, du point B au point A ; une partie du profil du sol est plus sèche (probablement en raison de l’effet combiné de l’évapotranspiration et de l’absorption de l’eau dans cette partie du sol par les racines et les organismes du sol).
Exemple d’étude de sol no 5 – Transport des solutés/lessivage des nitrates
Fernanda, étudiante à l’Université de la Saskatchewan, a mené une expérience sur la rétention d’eau à partir d’un dispositif constitué d’une colonne remplie de sol rattachée à un bassin de référence. Le diamètre la colonne est de 20 cm. Un dispositif installé au haut de la colonne laissait passer une solution de NO3- à un débit constant. Une fois que le débit de solution est devenu régulier au bas de la colonne (débit constant), Fernanda a collecté 30 ml de la solution en 30 minutes.
A) Calculez le débit d’eau dans le sol (débit massique d’eau)
Le flux d'eau est $q_w = \dfrac{V}{At}$ où $A$ est l'aire de la section transversale du poteau ($\pi r^2$); thus,
$q_w = \dfrac{30\,cm^3}{3.14\cdot (10\,cm)^2\cdot 30\,min} = 0.00318\,cm\,min^{-1}$
Fernanda a également prélevé un échantillon de la solution recueillie au bas de la colonne pour mesurer sa concentration en NO3- à l’aide de la chromatographie par échange d’ions. La valeur de la concentration de NO3- était égale à 10 μg L-1.
B) Calculez le débit massique de NO3-
Le flux de NO3- peut être calculé à l’équation n° 22 :
$q_s = q_wC = 0.00318\,cm\,min^{-1}\,\cdot\,10\,\mu g\,L^{-1}\cdot\,0.001\,L\,cm^{-3}$
$q_s = 3.18\cdot 10^{-5}\,\mu g \,cm^{-2}\,min^{-1}$
Sinon, il est possible de calculer le débit en fonction du flux chimique, défini comme étant :
$mass\,of\,chemicals = V \cdot C = 30 \,cm^3 \cdot 10 \,\mu g\,L^{-1} \cdot 0.001 \,L \,cm^{-3} = 0.3 \,\mu g$
ainsi,
$q_s = \dfrac{m}{At} = \dfrac{0.3\, \mu g}{3.14\cdot (10\,cm)^2\cdot 30\,min} = 3.18\cdot 10^{-5}\,\mu g\,cm^{-2}\,min^{-1}$
RÉFÉRENCES
Bittman, S., Sheppard, C., and Hunt, D. 2017. Potentiel d’atténuation de l’ammoniac atmosphérique au Canada. Utilisation et gestion des sols, 33 (2), 263-275. doi : 10.1111/ /résumé/12336
Caron, J., Pelletier, V., Kennedy, C. D., Gallichand, J., Gumiere, S., Bonin, S., Bland, B., and Pepin, S. 2017. Lignes directrices des stratégies de gestion de l’irrigation et du drainage pour l’amélioration de la production de canneberges et de l’optimisation de l’utilisation de l’eau en Amérique du Nord. Revue canadienne des sciences du sol, 97(1): 82-91.
Caron, J., Price, J. S., and Rochefort, L. 2015. Physical properties of organic soil: adapting mineral soil concepts to horticultural growing media and Histosol characterization. Vadose Zone Journal 14:1-14.
De Vries, D.A. 1963. Thermal properties of soils. In W.R. van Wijk (ed.) Physics of Plant Environment, John Wiley & Sons, New York. pp. 210-235.
Gregorich, E.G., Turchenek, L.W., Carter M.R., and Angers D.A. 2001. Soil and environmental science dictionary. CRC Press, Boca Raton.
Krzic, M., Wiseman, K., Dampier, L., Grand, S., Wilson, J. and Gaumont-Guay, D. 2013. SoilWeb200: An Online Educational Tool for the APBI 200 course: Introduction to Soil Science. The University of British Columbia, Vancouver [http://soilweb200.landfood.ubc.ca]
Narasimham, T. N. 2007. Central ideas of Buckingham (1907): A century later. Vadose Zone Journal 6(4):687-693. doi:10.2136/vzj2007.0080
Reicosky, D. C., and Ritchie, J. T. 1976. Relative importance of soil resistance and plant resistance in root water absorption. Soil Science Society of America Journal 40(2):293-297.
Rekika, D., Caron, J., Rancourt, G. T., Lafond, J. A., Gumiere, S. J., Jenni, S., and Gosselin, A. 2014. Optimal irrigation for onion and celery production and spinach seed germination in Histosols. Agronomy Journal 106(3):981-994.
Ritchie, H., and Roser M. 2013. Crop Yields. Published online at OurWorldInData.org retrieved from https://ourwolrdindata.org/crop-yields.
Tisdale, J.M. and Oades, J.M. 1982. Organic matter and water-stable aggregates in soils. European Journal of Soil Science 32(2):141-163. doi.org/10.1111/j.1365-2389.1982.tb01755.x
USD-NRCS. 1991. Soil-Water-Plant Relationships. Section 15, Chapter 1, National Engineering Handbook, 2nd edition. https://directives.sc.egov.usda.gov/OpenNonWebContent.aspx?content=18350.wba
Les Auteurs
Sandra Brown, professeure adjointe en enseignement, Faculté des systèmes paysagers et agro-alimentaire de l'Université de la Colombie-Britannique, Vancouver, BC

Peu de temps après l'éruption du mont Saint Helens en 1980, je me suis retrouvée dans la zone rouge - curieusement si grise, si sombre, si sinistre, et pourtant très belle. Un an plus tard, le paysage avait déjà commencé à se transformer. J'ai été fascinée, je voulais en savoir plus sur la transformation des cendres en sol. Les andisols (sols formés à partir des cendres volcaniques) sont mes préférés, en partie en raison de leur capacité à retenir l'eau, et en partie parce qu'ils sont parfaits pour la culture du café colombien!
Asim Biswas, professeur agrégé, École des sciences de l'environnement, Université de Guelph, Guelph, ON

Pour avoir grandi dans une petite ferme de subsistance dans un village isolé en Inde, je me suis vite rendu compte à quel point le sol était la ressource la plus précieuse de la ferme. Enfant, tout en jouant dans la terre ou en aidant les plus vieux aux cultures, je demeurais fasciné par le phénomène qui faisait qu'une toute petite graine plantée dans le sol puisse produire une grosse plante, voire un arbre ! Cette fascination m'a mené vers des études universitaires en sciences du sol. Mes recherches actuelles sont centrées sur l'augmentation de la productivité et de la résilience de notre production alimentaire dans la perspective de l'environnement durable en agriculture.
Jean Caron, professeur, Sciences du sol et génie agroalimentaire, Université Laval, QC Québec

J'avais 12 ans quand j'ai lu un article dans le Reader's Digest sur le célèbre agronome américain, Norman Borlaug. Il ne m'en fallait pas plus pour décider de devenir agronome à mon tour, décision que je n'ai jamais regrettée. Mon intérêt manifeste pour les mathématiques, la physique et la technologie m'ont naturellement mené à me spécialiser en physique du sol. À titre de professeur, mon enseignement couvre large: mouvement de l'eau dans les sols, gestion du drainage et de l'irrigation et conservation des sols. La théorie que j'enseigne trouve tout son sens quand, à titre de chercheur en recherche appliquée, je participe à la résolution de problèmes rattachés à la production agricole. Quelle opportunité que de pouvoir travailler avec des agriculteurs, ingénieurs agricoles et autres professionnels aussi passionnés que moi.
Miles Dyck, professeur agrégé, Faculté des sciences agricoles, de la vie et de l'environnement, Université de l'Alberta, Edmonton, AB

J'enseigne la physique du sol, la pédogénèse et la classification des sols. L'influence des activités humaines sur les processus physiques et les propriétés du sol constitue l'essentiel de mes recherches. J'ai toujours été curieux de comprendre l'évolution des pédo-paysages entourant la ferme où j'ai grandi, dans le sud-ouest de la Saskatchewan. Ce que j'ai appris sur les sols du Canada m'a simplement rendu accro! Je me sens privilégié d'avoir comme gagne-pain l'acquisition des connaissances sur les sols, si essentiels à la production alimentaire et à la qualité de l'eau et de l'air.
Bing Si, professeur, Collège d'agriculture, Université de la Saskatchewan, Saskatoon, SK

Quand j'étais adolescent, j'aimais aider ma mère aux travaux de ferme, notamment au hersage du sol. Mais après une longue journée d'hersage, je constatais bien que le sol demeurait toujours aussi dur qu'une roche. C'était un sol argileux, faible en matière organique, et quand il était sec, une croûte dure se formait en surface. C'est de ce problème que je souhaitais résoudre que découle ma décision de m'inscrire à une majeure en agriculture à l'université. Mes recherches actuelles portent sur les sols sableux. Je cherche la raison pour laquelle ces derniers peuvent soutenir une forêt plus exigeante en eau et non une culture. Et j'étudie aussi la part que la profondeur du sol (de plus de 1 m) joue dans la croissance des plantes.
