Se creuser la tête
5 La chimie du sol
Darshani Kumaragamage; Jim Warren; and Graeme Spiers
OBJECTIFS D’APPRENTISSAGE
À la fin de ce chapitre, l’étudiant devrait être en mesure :
- de décrire les propriétés chimiques du sol et l’importance de leur contribution au maintien de la vie sur terre
- de décrire les structures des colloïdes organiques et inorganiques du sol, leurs propriétés et le phénomène d’apparition des charges
- de décrire en quoi la composition en minéraux dans un sol est déterminante sur certaines de ses propriétés
- d’avoir assimilé les notions de capacité d’échange cationique et d’adsorption anionique ; de connaître les facteurs qui influencent la capacité d’échange d’ions dans le sol
- de déterminer les facteurs qui régissent les gradients d’acidité et d’alcalinité dans le sol, de reconnaître les caractéristiques d’un sol fortement acide et d’un sol fortement alcalin et des effets que ces caractéristiques exercent sur la croissance des plantes
- de connaître les causes de salinité d’un sol, de définir les caractéristiques des sols salins, des sols salins-sodiques et des sols sodiques ; d’en savoir plus sur les possibilités de réhabilitation des sols touchés par la salinisation
- d’expliquer en quoi les réactions d’oxydoréduction sont importantes dans la dynamique des éléments nutritifs et des émissions de gaz à effet de serre
- de comprendre les principes à la base des mesures de (1) la capacité d’échange cationique, (2) du pH, (3) de la conductivité électrique et (4) du potentiel d’oxydoréduction et d’avoir appris à interpréter les valeurs de ces mesures
INTRODUCTION
La chimie du sol est la branche de la science du sol qui traite de la composition chimique, des réactions chimiques et des propriétés chimiques des sols (Sparks et coll., 2019). Les composantes non biotiques du sol comprennent les trois phases de la matière, soit les solides (matériaux organiques et inorganiques [minéraux]), les liquides (l’eau du sol) et les gaz (l’air du sol) ; sa composante biotique est constituée des organismes vivants. Cet environnement physico-chimique spécifique associé à la communauté des organismes qui y vivent constitue un écosystème dans lequel les ions et les molécules en présence se déplacent d’une de ses composantes à l’autre du fait de leurs interactions constantes.
On caractérise les propriétés chimiques du sol à partir de mesures des concentrations et des proportions d’espèces chimiques, soit dissoutes dans l’eau du sol, soit présentes sur le complexe d’échange d’ions. Les propriétés chimiques du sol, telles que la capacité d’échange cationique (CEC), le pH, le potentiel d’oxydoréduction (Eh) ou pe, et la conductivité électrique (CE) exercent une influence déterminante sur la disponibilité des éléments nutritifs, la croissance des plantes, le comportement des polluants, l’activité biologique, etc. La CEC est la quantité totale de cations (ions chargés positivement) qu’une surface minérale ou organique chargée négativement peut adsorber. La CEC est le plus souvent exprimée en centimoles de charge [cmol (+)] par kg de sol. Le pH du sol est une mesure de la concentration de H+ dans la solution de sol, tandis que l’Eh fournit un indice de la concentration des espèces chimiques d’éléments sensibles à l’oxydoréduction, par exemple, le fer (Fe3+ / Fe2+). La conductivité électrique (CE) fournit une indication de la quantité de sels solubles dans le sol.
Les propriétés chimiques du sol jouent un rôle prépondérant dans la production agricole, la protection de la sécurité alimentaire et la biodiversité. En effet, à partir de ses propriétés chimiques, il devient possible d’évaluer dans quelle mesure un sol peut subvenir non seulement aux besoins des plantes, mais aussi à tous les autres organismes. Par exemple, étudier la manière dont les éléments nutritifs (azote, calcium, phosphore) de même que les contaminants (métaux lourds, pesticides) réagissent et circulent dans le sol permet de prévoir sa capacité à poursuivre son rôle indispensable de milieu de vie. Par exemple, la simple présence d’éléments nutritifs ou de contaminants en provenance du sol retrouvés dans un cours d’eau (sous forme soluble) ou dans l’atmosphère (sous forme gazeuse) peut devenir source de pollution des eaux de surface ou souterraines, de l’air, de même que de contribuer au réchauffement climatique, à l’appauvrissement de l’ozone stratosphérique, etc.
Les réactions chimiques qui se produisent par contact entre l’eau du sol (dite solution de sol) et sa fraction colloïdale sont révélatrices de la nature des propriétés chimiques qui caractérisent un sol donné. La fraction colloïdale comprend les surfaces de la matière organique et de la matière minérale (particules <0,002 mm de diamètre ; Brady et Weil, 2010).
La première section de ce chapitre traite de la chimie de la solution de sol et de sa fraction colloïdale. Les autres sections présentent les principales propriétés et caractéristiques chimiques du sol, soit la capacité d’échange cationique (CEC), la capacité d’échange d’anions, le pH du sol, la conductivité électrique (CE) et le potentiel d’oxydoréduction (Eh), envisagées dans leur contribution fondamentale au maintien de la vie sur terre.
SOLUTION DE SOL
On appelle communément « solution de sol » l’eau et les solutés dissous qu’un sol contient. La solution de sol en est l’élément moteur. En effet, la solution de sol sert à la fois de milieu où se produisent les réactions chimiques des diverses substances présentes dans le sol et de voie de circulation de ces substances. Quelques exemples de réactions chimiques comptent les réactions de précipitation/dissolution des minéraux, les échanges d’ions, les réactions d’oxydoréduction, l’absorption des éléments nutritifs par les plantes, etc. L’importance de la présence d’eau dans le sol est telle que s’il n’y en avait pas, le peu de réactions chimiques et biologiques qui s’y produiraient serait insuffisant pour assurer la vie sur terre.
Dans la solution de sol, on trouve une grande variété de cations et d’anions (à la fois sous forme d’ions libres et d’ions complexes) ainsi que des molécules organiques dissoutes, généralement à de faibles concentrations. Pour que les racines des plantes puissent absorber les éléments nutritifs, ces derniers doivent se trouver dissous dans la solution de sol. On estime qu’il n’y a pas moins de 17 éléments qui sont nécessaires à la croissance de la plupart des plantes (voir chapitre 7); certaines autres auraient besoin d’un supplément de 4 éléments (Havlin et coll., 2013). Les cations et anions que les plantes absorbent de la solution du sol comptent : l’azote sous forme d’ammonium (NH4+) et de nitrate (NO3–), le phosphore sous forme de plusieurs espèces de phosphate (p. ex. HPO42- et H2PO4–), le potassium (K+), le calcium (Ca2+), le magnésium (Mg2+) et le soufre sous forme de sulfate (SO42-). Tous les éléments nutritifs que les plantes absorbent par les racines proviennent de la solution de sol.
La solution de sol contient aussi d’autres ions et molécules qui ne sont d’aucune utilité pour les plantes. Certains de ces autres ions et molécules ne présentent aucune toxicité pour elles, alors que d’autres le sont en fonction de leur concentration. Ces derniers comprennent certains oligo-éléments (p. ex. plomb, arsenic), pesticides (p. ex. chlorpyrifos, glyphosate), ions et composés antimicrobiens (p. ex. désinfectants, antibiotiques) et certaines molécules organiques complexes. Toutes ces substances sont susceptibles d’être absorbées par les plantes. Ces mêmes substances présentes dans la solution du sol peuvent également être évacuées du sol par ruissellement de surface ou par lessivage, en particulier en cas de précipitation ou d’irrigation excessives.
COLLOÏDES DU SOL
La fraction colloïdale du sol comprend la fraction du sol constituée des petites particules inorganiques et organiques (<0,002 mm) (Brady et Weil, 2010). La plupart de ces particules sont dotées d’une grande surface spécifique (par rapport à leur taille) et sont porteuses d’une charge négative. Ces deux caractéristiques confèrent au sol d’importantes capacités, telles que celle de gonfler, de retenir l’eau et de la faire circuler, d’adsorber des cations et des anions et d’échanger des ions.
La capacité des colloïdes du sol de pouvoir interagir avec la solution de sol tient en grande partie à ce rapport particulier qui existe entre leur grande surface spécifique et leur taille relativement petite. Le tableau 5.1 montre le rapport entre la taille (ou dimension ou grosseur) des particules d’un loam argileux (groupées suivant les trois classes de la fraction de terre fine – sable, limon, argile) et leur surface spécifique. La surface spécifique d’une particule solide définit sa surface par unité de masse. On l’exprime en m2 g-1 de sol. Comme on peut le constater dans le tableau, ce sont les particules de la classe des argiles (<2 µm) qui offrent le plus de surface spécifique (99 % en termes relatifs) par rapport aux particules des deux autres classes qui composent ce loam argileux, même si les trois classes sont présentes en proportions égales.
Tableau 5.1. Relation entre la taille des particules solides d’un loam argileux (groupée en classes) et leur surface spécifique
| Classe de la fraction fine du sol |
Limites des classes |
Diamètre médian |
Composition du loam argileux a |
Surface spécifique b | Surface spécifique totale |
|---|---|---|---|---|---|
| (µm) | (µm) | (%) | (cm2 g-1) | (%) | |
| Sable | 2000-50 | 1000 | 33.3 | 22.6 | 0.2 |
| Limon | 50-2 | 25 | 33.3 | 90.9 | 0.8 |
| Argile | <2 | 1 | 33.3 | 11,320 | 99 |
| a On suppose un loam argileux constitué de proportions égales de sable, de limon et d’argile. b Calculé sur le modèle du diamètre médian de particules sphériques uniformes. | |||||
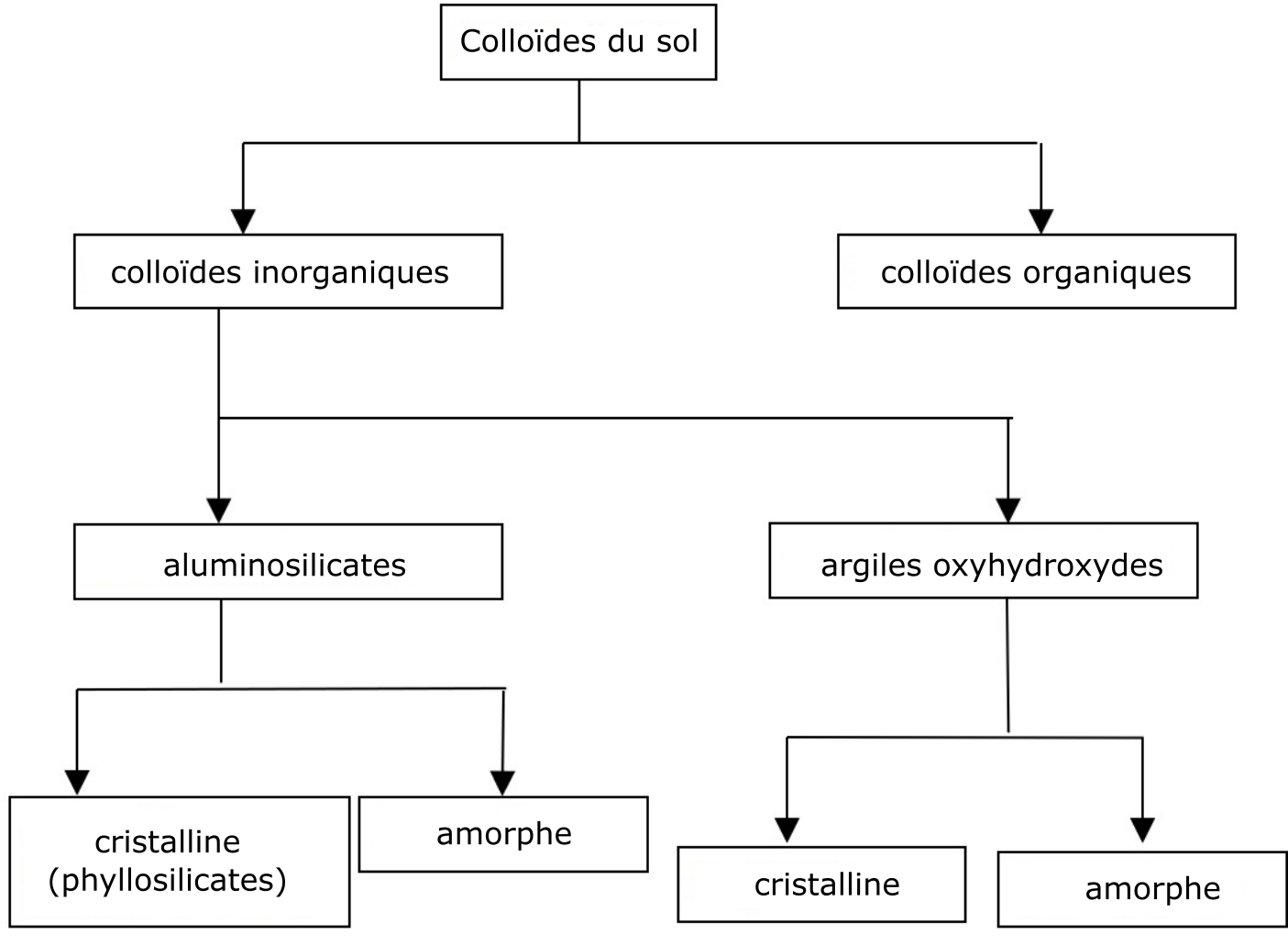
Les particules minérales de la fraction colloïdale ou argileuse les plus abondantes sont les phyllosilicates et les oxyhydroxydes (Figure 5.1). Les particules organiques de la fraction colloïdale (colloïdes organiques) sont à peu près de la même taille que celle des particules minérales. Les colloïdes organiques sont constitués de petites particules d’humus relativement stables, résistantes à la biodégradation. Les particules colloïdales peuvent exister telles quelles dans l’environnement du sol ou se trouver sous forme de complexes organo-minéraux (Newman et Hayes, 1990).
Phyllosilicates
Les colloïdes minéraux (aussi appelés « colloïdes d’argile ») les plus abondants sont ceux qui appartiennent au groupe de composés appelé « silicates d’aluminium » (aussi appelés « phyllosilicates » ou « silicates stratifiés »). Les différences marquées qui existent entre chacun de ces composés de silicates d’aluminium se reflètent dans leurs propriétés respectives, par exemple dans la capacité d’échange d’ions.
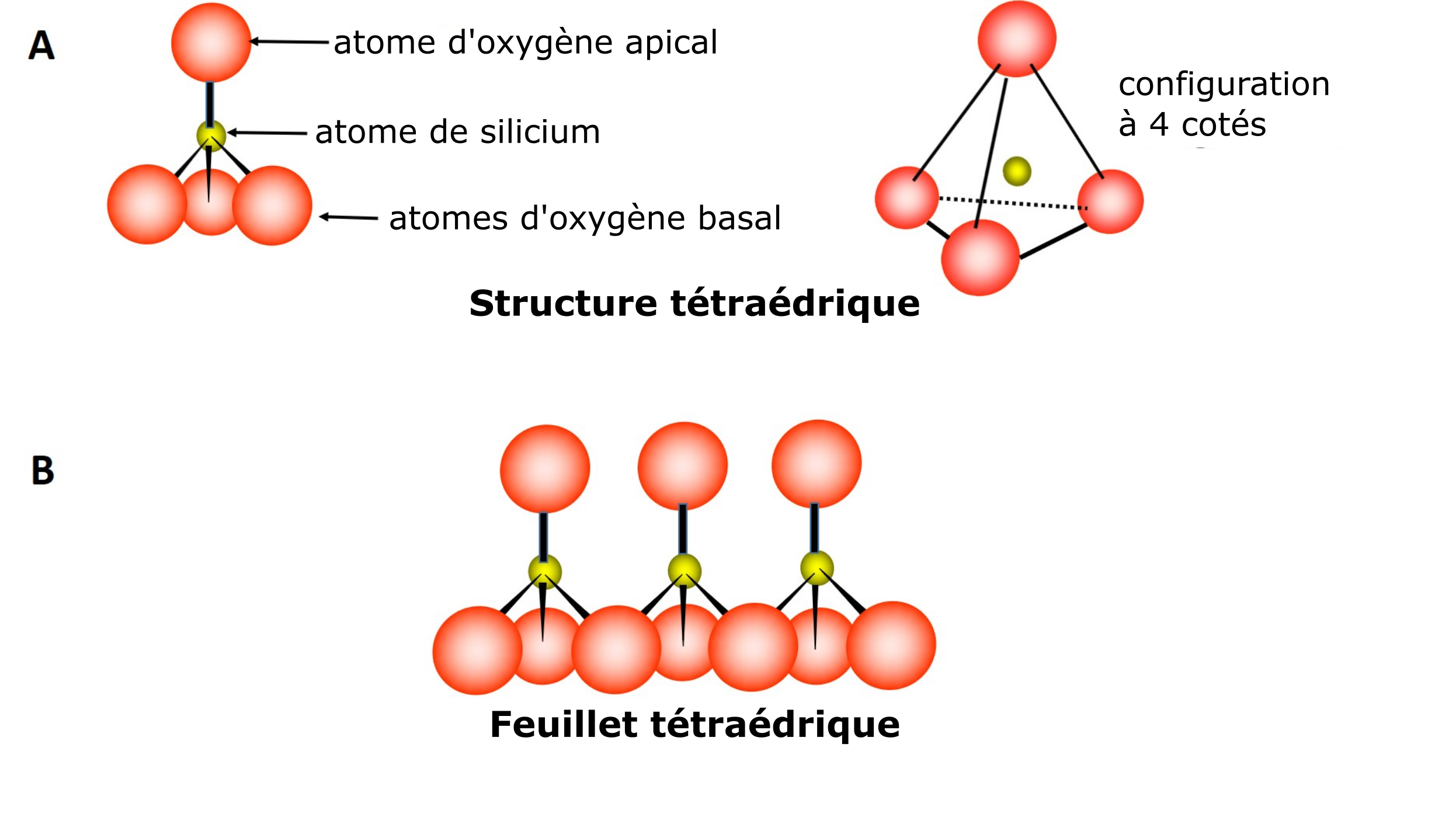
Les phyllosilicates (phyllon qui signifie feuille ou feuillet en grec) appartiennent à la classe des minéraux cristallins silicatés. Les silicates sont des minéraux formés de feuillets (feuilles, lamelles) constitués d’unités atomiques identiques. Deux types d’unités atomiques peuvent former un feuillet, qui se distinguent par leur structure et leur composition chimique. La première unité est caractérisée par une structure moléculaire tétraédrique composée de quatre atomes d’oxygène et d’un atome de silicium. La structure tétraédrique est donnée par la disposition en apex d’un atome d’oxygène par rapport à l’atome de silice et des trois autres à sa base (Figure 5.2A). La formation d’un feuillet résulte du partage des trois atomes basaux d’une unité tétraédrique avec trois atomes de l’unité tétraédrique voisine, et ainsi de suite (Figure 5.2B).
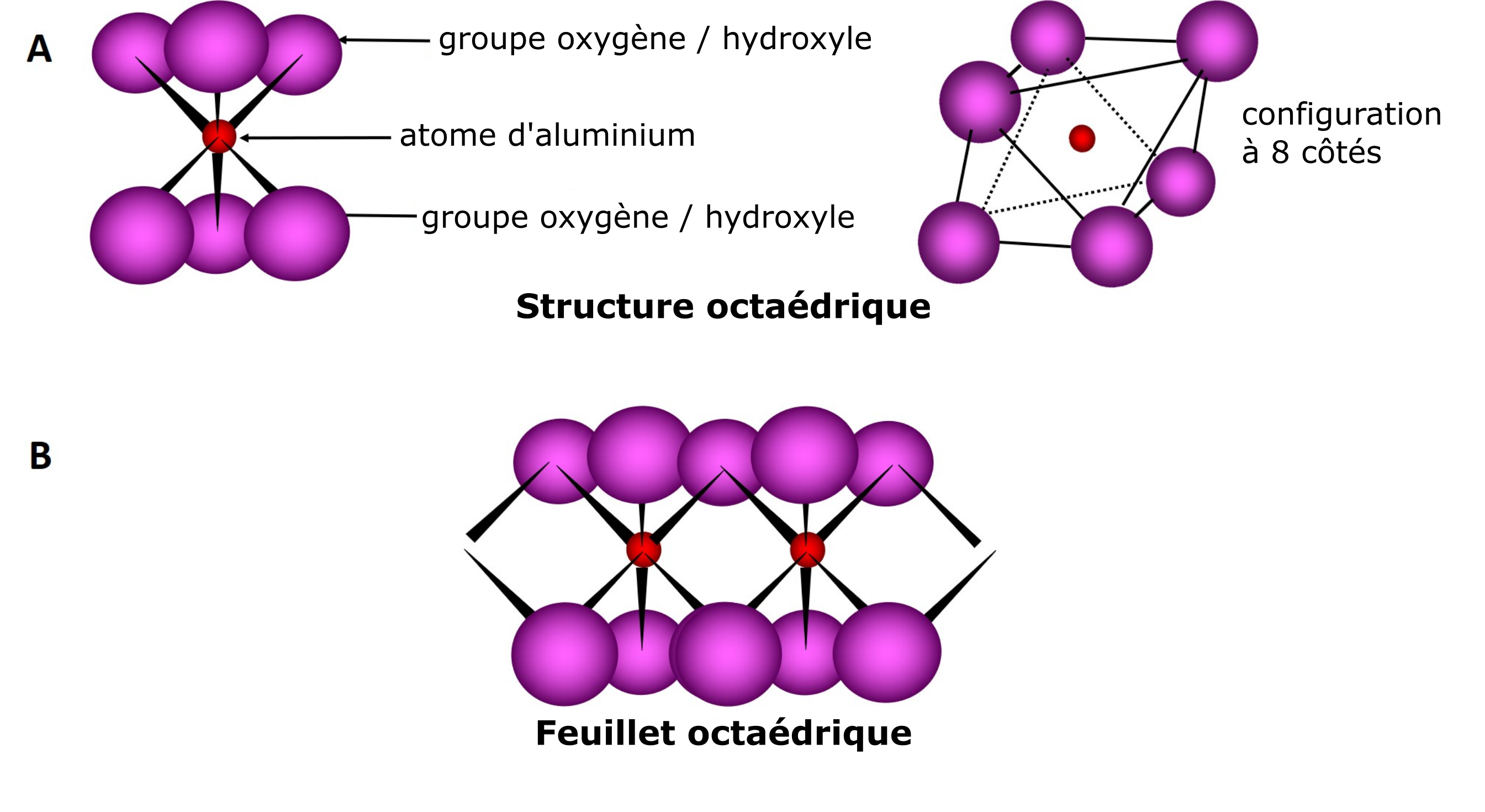
La deuxième unité est caractérisée par une structure moléculaire octaédrique qui est composée d’atomes d’oxygène ou d’hydroxyles (aussi appelés « groupes hydroxyles » ou « groupes-OH »), lesquels entourent l’un ou l’autre des cations Si, Al, Mg, etc. La structure octaédrique est donnée par la disposition des six atomes d’oxygène ou d’hydroxyles situés également de part et d’autre du cation. La formation d’un feuillet résulte du partage des atomes d’oxygène d’une unité octaédrique avec les atomes d’oxygène de l’unité octaédrique voisine et ainsi de suite (Figure 5.3).
Les feuillets tétraédriques et octaédriques se superposent en couches de différents phyllosilicates par le partage de l’atome d’oxygène apical du feuillet tétraédrique avec des atomes d’oxygène du feuillet octaédrique. Les phyllosilicates composés d’un feuillet tétraédrique et d’un feuillet octaédrique sont appelés « phyllosilicates de type 1:1 » ; ceux composés d’un feuillet octaédrique pris en sandwich entre deux feuillets tétraédriques sont appelés « phyllosilicates de type 2:1 » (Figure 5.4). La composition « théorique » de l’unité de base d’un minéral argileux de type 1:1 est Si4Al4O10(OH)8, tandis que celle d’un minéral argileux de type 2:1 est Si8Al4O20(OH)4.
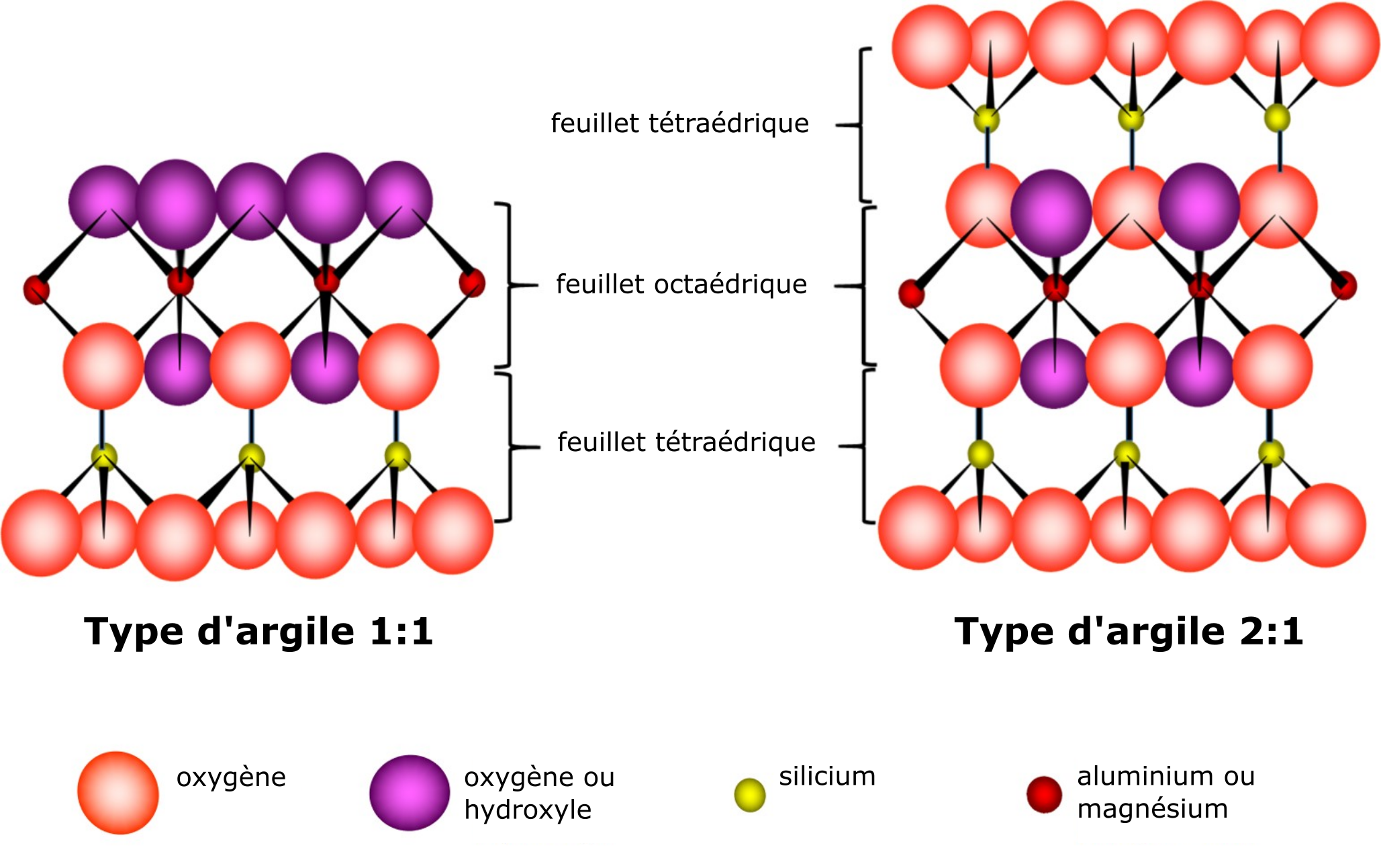
Les argiles phyllosilicates sont formées de nombreuses couches soit du type de phyllosilicate 1:1 soit du type de phyllosilicate 2:1 empilées les unes sur les autres et retenues entre elles par diverses formes de liaisons. La position intercouche définit l’espace entre deux couches. Les caractéristiques associées à ces espaces entre les couches des deux types de phyllosilicates (1:1 ; 2:1) déterminent en grande partie leur comportement chimique de base et leurs propriétés chimiques.
Substitution Isomorphe
En plus d’être dotés d’une grande surface (voir tableau 5.1), les phyllosilicates sont également porteurs d’une charge électrique négative. Pour cette raison, les cations constitutifs des feuillets tétraédriques ou octaédriques peuvent par substitution être remplacés par d’autres cations de taille semblable, mais de valence différente (voir tableau 5.2).
On définit la substitution isomorphe comme désignant « le remplacement d’un atome par un autre de taille similaire dans une structure cristalline sans perturber ni modifier la structure cristalline du minéral » (ministère de l’Agriculture du Canada, 1976). Dans le cas où un cation de substitution a une valence moindre que celui qu’il remplace, les atomes d’oxygène qui l’entourent deviennent porteurs d’une charge négative, conférant une charge négative nette à toute la structure du feuillet. Par exemple, l’aluminium trivalent (Al3+) peut se substituer à certains des atomes de silicium tétravalents (Si4+) dans un feuillet à structure tétraédrique. De même, dans un feuillet à structure octaédrique, Mg2+, Fe2+, ou d’autres cations divalents peuvent se substituer à certains Al trivalents (Al3+), apportant au feuillet un supplément de charge négative. Les structures tétraédrique ou octaédrique chargées négativement attirent des cations chargés positivement. La principale source de cations neutralisateurs de la charge négative des phyllosilicates se trouve entre les couches de phyllosilicates du type 1:1 ou du type 2:1. L’abondance des charges négatives (c.-à-d. la densité de charge), leur emplacement dans le feuillet tétraédrique ou octaédrique et les cations neutralisateurs (p. ex. Na+, Ca2+, Mg2+, K+ ou autres) situés entre les couches de ces types d’argiles déterminent la capacité des phyllosilicates à retenir leurs cations de même que le degré auquel ils se dilatent ou se gonflent.
Tableau 5.2. Rayon des cations les plus communs et position dans les structures phyllosilicates
| Cation | Rayon (nm) | Position |
|---|---|---|
| Si4+ | 0.041 | tructure tétraédrique |
| Al3+ | 0.05 | tructure tétraédrique, octaédrique et position intercouche |
| Fe3+ | 0.064 | octaédrique |
| Fe2+ | 0.076 | octaédrique et position intercouche |
| Mg2+ | 0.065 | octaédrique et position intercouche |
| Ca2+ | 0.099 | position intercouche |
| Na+ | 0.095 | position intercouche |
| K+ | 0.133 | position intercouche |
Minéraux Phyllosilicates
Il existe environ 50 espèces de minéraux phyllosilicates, mais cinq seulement sont communs dans les sols canadiens (voir chapitre 14). On trouve des illustrations de ces cinq minéraux phyllosilicates à la Figure 5.5. On les décrit ci-dessous.
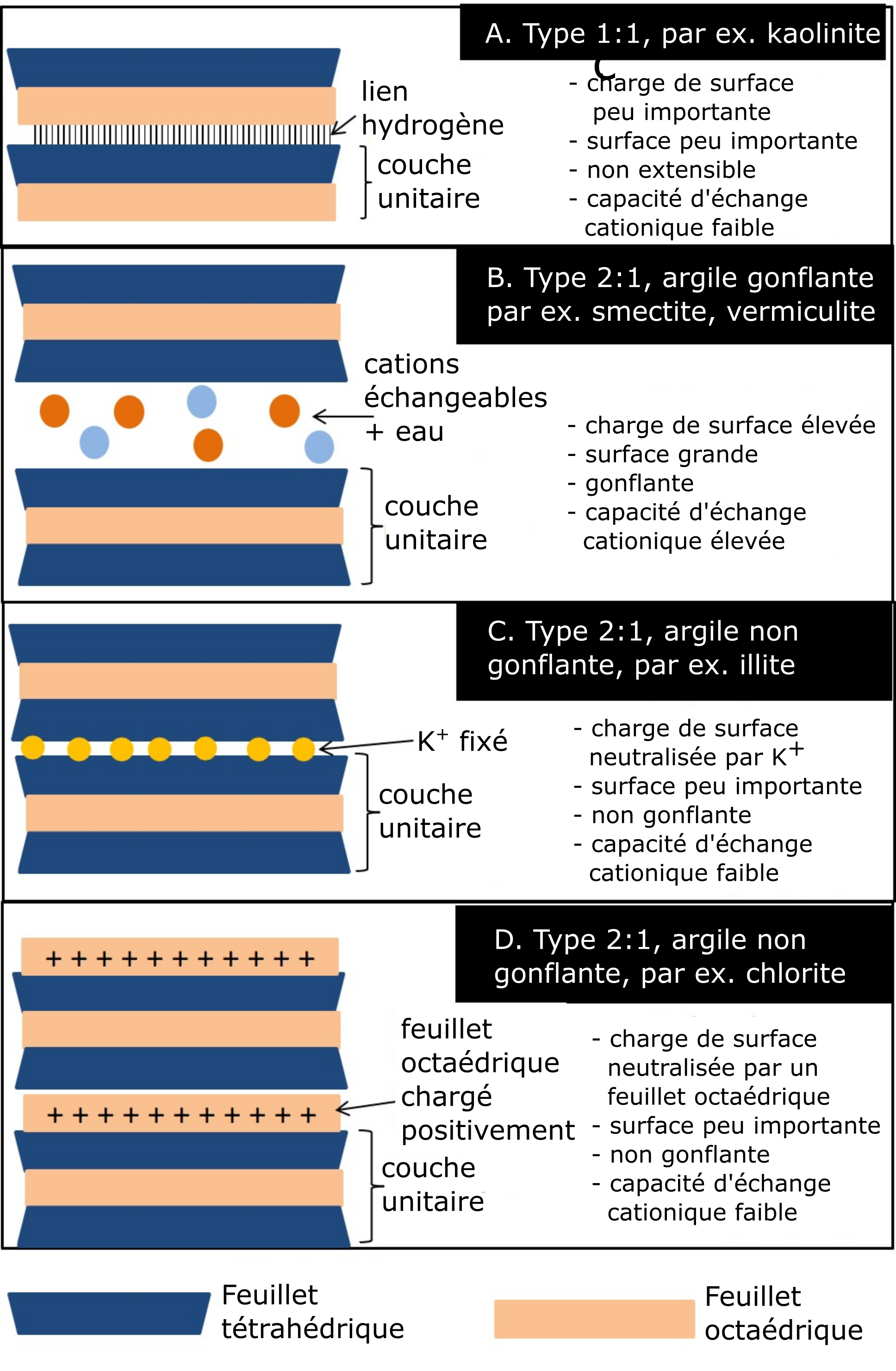
La kaolinite (Figure 5.5A) est une argile de type 1:1, dont les couches sont maintenues ensemble par des liaisons hydrogène. Une liaison hydrogène attache l’hydroxyle d’un feuillet octaédrique d’une couche aux oxygènes (basaux) du feuillet tétraédrique de la couche adjacente. Ces liaisons hydrogène tiennent très serrée la structure de cette argile la rendant ainsi très stable. Le caractère stable de cette argile de type 1:1 combiné au fait qu’elle est très peu sujette à la substitution isomorphe empêche les cations et l’eau de s’immiscer entre les couches. Du fait de ces deux caractéristiques, la kaolinite gonfle peu. En effet, les molécules d’eau ne pouvant pas s’immiscer entre les couches, ce minéral ne peut ni gonfler ni rétrécir. Parce que ses intercouches se trouvent non exposées, la kaolinite offre peu de surface spécifique. Compte tenu du fait qu’il ne peut y avoir de substitution isomorphe dans la kaolinite, sa charge négative de surface est faible et par le fait même, sa capacité d’échange cationique.
Matière à réflexion
La smectite (Figure 5.5B) est le nom d’un groupe de phyllosilicates de type 2:1 gonflante. La montmorillonite et la beidellite sont les deux espèces les plus communes du groupe des smectites dans les sols canadiens. La substitution isomorphe dans ces deux phyllosilicates a été modérée par rapport aux autres phyllosilicates de ce groupe (le mica hydraté, p. ex.). Ces deux espèces de minéraux phyllosilicates se distinguent l’un de l’autre par le lieu de substitution à l’intérieur du feuillet constitutif de la couche unitaire gonflante de type 2:1. Dans le cas de la montmorillonite, la substitution isomorphe s’est produite en majorité dans le feuillet octaédrique de la couche unitaire 2:1, tandis que dans le cas de la beidellite, elle a eu lieu dans le feuillet tétraédrique. Deux autres caractéristiques distinguent ce type de phyllosilicates : les feuillets tétraédriques qui se font face n’ont pas de groupes hydroxyle, d’où leur incapacité d’avoir des liaisons hydrogène ; la présence de six molécules d’eau agencées selon une configuration octaédrique entoure chacun des cations échangeables présents dans les intercouches. Les couches constituantes de la structure de ces phyllosilicates se trouvent seulement retenues par les forces électrostatiques neutralisantes des cations échangeables présents dans les intercouches. Ces forces tendent à neutraliser la charge négative résultant de la substitution isomorphe qui a eu lieu dans les feuillets de chaque couche unitaire. Dans les intercouches, chaque cation échangeable se trouve entouré de six molécules d’eau agencées selon une configuration octaédrique. La faiblesse de ces forces électrostatiques qui tiennent ensemble les couches facilite la pénétration des molécules d’eau et des cations. C’est ce qui donne à ce groupe sa propriété de gonflement/rétrécissement, une surface spécifique élevée et une capacité d’échange cationique élevée.
La vermiculite (Figure 5.5B) est également un phyllosilicate de type 2:1, mais dont la substitution isomorphe est intermédiaire (couche de charge négative) entre celle de la smectite et celle des phyllosilicates de mica hydraté / illite de type 2:1 (Figure 5.5C). De ce fait, la vermiculite se trouve limitée en matière de capacité gonflement/rétrécissement. En revanche, la vermiculite est dotée d’une charge de surface négative élevée et d’une capacité d’échange cationique élevée, étant donné que la plupart des substitutions isomorphes se sont produites dans les feuillets tétraédriques. Une telle composition des couches unitaires permet à cette structure de piéger dans ses intercouches les ions K+, comme dans le cas des phyllosilicates de mica hydraté/illite, mais comme la charge négative de ses couches est légèrement inférieure à celle des couches des micas hydratés/illite, le K piégé peut être relâché dans la solution du sol contrairement au mica hydraté/illite.
Le mica hydraté aussi appelé illite (Figure 5.5C) est un autre minéral argileux de type 2:1. Le mica hydraté est un dérivé de l’altération partielle du mica, un minéral primaire commun dans les roches granitiques du Bouclier canadien. La structure que forment les couches du mica/illite hydraté ressemble à celle des smectites et à celle de la vermiculite, à une exception près : chaque couche unitaire est porteuse d’une charge négative plus élevée qui résulterait de la substitution isomorphe presque exclusivement produite dans les feuillets tétraédriques, où Al3+ a été substitué à Si4+. Le potassium (K+) occupe presque exclusivement les intercouches, ce qui a pour effet de maintenir la structure par neutralisation de la charge négative des couches unitaires qui la constituent. L’ion potassium (K+) possède juste la bonne taille pour s’insérer dans les trous de forme hexagonale des feuillets tétraédriques des couches 2:1. Un tel rapprochement des couches chargées négativement crée des liens plus serrés entre les couches qui forment la structure de ce type 2:1 non gonflant. Ces liens étroits entre les couches empêchent l’eau d’entrer dans les intercouches, privant de ce fait le groupe des micas hydratés/illite de la capacité de gonflement. Ainsi, même si la charge négative des couches unitaires dépasse celle des smectites et celle de la vermiculite, la capacité d’échange cationique de ce groupe reste faible, car la charge négative se trouve neutralisée par les ions K+.
La chlorite (Figure 5.5D) est un groupe de phyllosilicates de type 2:1:1 (ou 2:2). Le minéral a une structure de type 2:1, mais dont les intercouches comprennent aussi un autre feuillet octaédrique – composé soit d’hydroxyde d’aluminium, soit de magnésium soit de fer agencé suivant une configuration octaédrique. Ce feuillet intercouche octaédrique ne partage pas ses molécules d’oxygène avec la couche unitaire 2:1, mais plutôt ses ions hydrogène du groupe des hydroxyles. En conséquence de quoi, le minéral se trouve privé de propriété de gonflement. La structure de la chlorite présente une variabilité de substitution isomorphe, mais ses charges négatives se trouvent neutralisées par le feuillet octaédrique chargé positivement, de sorte que sa capacité d’échange d’ions demeure faible.
Les aluminosilicates amorphes sont d’autres aluminosilicates que l’on trouve dans le sol qui se distinguent des phyllosilicates (voir Figure 5.1) par leur structure. Ces aluminosilicates amorphes se présentent en série rapprochée (short-range orderered aluminosillicates) par allusion à leur structure qui n’est pas constituée de feuillets unitaires comme les phyllosilicates. L’allophane et l’imogolite sont deux exemples d’aluminosilicates amorphes. De tels minéraux se trouvent en abondance au Japon et en Nouvelle-Zélande dans les sols d’origine volcanique. On pourra en trouver au Canada en très petites quantités dans certains podzols et solonetzs. Ces aluminosilicates amorphes n’ont pas la propriété de pouvoir gonfler. Ils sont extrêmement petits (<4 nm de diamètre), d’une surface spécifique très élevée et d’une capacité d’échange d’ions également très élevée.
Minéraux Oxyhydroxydes
Les minéraux oxyhydroxyde (sesquioxydes) sont principalement des oxydes, des hydroxydes et des oxydes hydratés de Fe, d’Al et de Mn, qui font partie des minéraux les plus récemment formés. Certains présentent une structure interne cristalline bien définie (p. ex. gibbsite, hématite, goethite), d’autres sont sans structure, c’est-à-dire amorphe ou non cristalline (p. ex. ferrihydrite). Les argiles à oxyde cristallin sont composées de groupes oxygène ou hydroxyle, qui forment une structure octaédrique avec le Fe, l’Al ou le Mn. Les structures octaédriques de ces minéraux sont maintenues par des liaisons H. Les oxyhydroxydes non cristallins (amorphes) résultent de la précipitation rapide du Fe, de l’Al et du Mn dans la solution de sol en réaction aux cycles de mouillage et de séchage. Un précipité se forme quand, dans la solution de sol, les cations (Al, Fe, Mn) et les anions (OH) se trouvent en excès (sursaturation). Le phénomène de précipitation peut aussi entraîner le piégeage dans la structure du précipité d’autres solutés comme le carbonate, le phosphate et le sulfate. Les oxyhydroxydes amorphes sont métastables, mais finissent par se convertir graduellement en une forme cristalline.
Les minéraux oxyhydroxydes, qu’ils soient cristallins ou amorphes, ne sont pas porteurs d’une charge négative permanente, bien qu’ils aient une charge de surface négative élevée associée à leurs groupes hydroxyle de surface. La capacité d’échange cationique des oxyhydroxydes dépend beaucoup du pH. Dans des conditions de faible pH, les protons se fixent (protonation) sur les groupes OH, les chargeant positivement ; dans des conditions de pH élevé, les groupes OH se dissocient, les rendant porteurs d’une charge négative. Or, on dit des substances qui revêtent cette capacité de pouvoir être autant porteuses d’une charge nette négative que positive qu’elles sont amphotères. Ainsi, à pH élevé, les minéraux oxyhydroxydes expriment leur capacité d’échange de cations, alors qu’à pH faible, c’est leur capacité d’échange d’anions qui se manifeste.
Les oxyhydroxydes se trouvent en petites quantités dans les sols canadiens (pédogénèse récente), mais on en trouve beaucoup plus fréquemment dans les sols très altérés des régions chaudes et humides dans les autres parties du monde. Ces minéraux très réactifs dotés d’une surface spécifique très élevée exercent une forte influence sur les propriétés du sol, telles que l’adsorption et l’échange d’ions.
Colloïdes Organiques (Humus)
Les colloïdes organiques, plus connus sous le nom d’humus, résultent de la décomposition de la matière organique. Ces colloïdes présentent une structure moléculaire très complexe, constituée d’un mélange de polymères organiques. Ces derniers sont principalement formés des éléments C, H et O. De tous les aspects de la chimie du sol, celui de ces composés est le moins compris. Toutefois, certaines de leurs propriétés sont bien connues, telles que leur réactivité chimique élevée et leur capacité de retenir et d’absorber l’eau. De plus, comparativement aux colloïdes inorganiques, les colloïdes organiques sont dotés d’une plus grande surface spécifique et d’une plus grande charge de surface. Les charges de surface principalement négatives et dépendantes du pH résultent de la dissociation partielle des groupes hydroxyle (-OH), carboxyle (-COOH) et phénoliques, lesquels sont associés à des structures de carbone centrales qui varient en taille et en complexité. Par unité de masse, si les colloïdes organiques ont une plus grande influence sur les propriétés du sol que les colloïdes inorganiques, leur présence dans le sol est en revanche moins importante
Charges sur les Colloïdes du Sol
Les charges dont sont dotés les colloïdes du sol résultent de deux mécanismes :
- Par substitution isomorphe dans la structure cristalline des phyllosilicates – qui conduit à la formation de charges permanentes, la plupart négatives. On se rappellera que la substitution isomorphe définit le phénomène du remplacement d’un atome par un autre de taille similaire dans une structure cristalline sans ni la perturber ni la modifier. Le remplacement d’un cation de plus grande valence par un cation de moindre valence (p. ex. substitution de Al3+ par Mg2+ dans le feuillet octaédrique) se trouve à charger négativement les surfaces minérales de façon permanente, indépendantes aux variations de pH.
- Par protonation / déprotonation des groupes fonctionnels à la surface des argiles oxyhydroxydes, sur les bords des argiles phyllosilicates et des composés organiques, qui donne lieu à l’apparition de charges positives ou négatives appelées « charge (variable) dépendante du pH ». La protonation des groupes fonctionnels se produit dans des conditions acides (pH bas), tandis que la dissociation des protons hydroxyles prédomine dans des conditions basiques (pH élevé) (Figure 5.6). Les conditions de pH élevé favoriseront l’apparition de charges négatives sur les groupes fonctionnels, qui deviendront principalement chargés positivement dans des conditions de bas pH. Le pH auquel les groupes fonctionnels des colloïdes du sol portent autant de charges positives que négatives est appelé « point de charge protonique nette zéro » ou pour faire plus court « point de charge zéro ». Tout comme les colloïdes inorganiques, les colloïdes organiques (l’humus) ont aussi en surface des groupes fonctionnels (p. ex. carboxyle, phénolique) qui peuvent se dissocier (déprotoner) en fonction du pH, ce qui leur confère une charge négative. Et à mesure que le pH augmente, le phénomène de dissociation sur les groupes fonctionnels augmente aussi, les rendant du coup encore plus chargés négativement. Les conditions de sol acide (pH bas) défavorisent la dissociation des groupes fonctionnels de sorte que la charge négative de surface des colloïdes organiques demeure faible.

Influence des colloïdes du sol sur les propriétés du sol
La quantité et les types de colloïdes inorganiques et organiques qui se trouvent dans le sol exercent une profonde influence sur ses propriétés. Par exemple, les sols contenant une forte proportion d’humus ont une capacité de rétention d’eau élevée. Ceux contentant une forte proportion de minéraux argileux gonflants, comme la smectite, rétrécissent en temps de sécheresse et gonflent en présence d’humidité. Les vertisols, sols à texture fine contenant > 60 % d’argile dont plus de la moitié est de la smectite, sont généralement très collants lorsqu’ils sont humides et traversés de crevasses profondes lorsqu’ils sont secs.
Le fait que les phyllosilicates, les oxyhydroxydes (des colloïdes inorganiques) et les colloïdes inorganiques (l’humus) soient dotés de capacité d’ionisation à leur surface, ils se trouvent à contribuer à la capacité d’ionisation totale des sols, et donc à la capacité d’échange cationique. On présente au Tableau 5.3 la capacité d’échange cationique (CEC) de quelques colloïdes du sol. Par exemple, un sol qui contiendrait 40 % d’argile smectite (avec une CEC de 100 cmol(+) kg-1) pourrait contribuer jusqu’à 40 cmol(+) kg-1 à la CEC totale du sol. Compte tenu de sa densité de charge élevée, l’humus peut être un contributeur important à la CEC totale, même dans les sols minéraux, en particulier dans des conditions de pH neutre à élevé, qui font dominer les charges négatives dépendantes du pH. Selon les caractéristiques de l’humus, le pH et les interactions qui ont lieu entre l’humus et d’autres particules du sol, une teneur en humus de 3 % (en supposant une CEC de 400 cmol(+) kg-1) peut contribuer à la CEC du sol jusqu’à 12 cmol(+) kg-1. Par ailleurs, la contribution à la CEC des argiles oxyhydroxyde de fer est également fonction du pH. Toutefois, la contribution de ces argiles à la CEC du sol est négligeable par rapport à la contribution de l’humus et des phyllosilicates à CEC élevée. En revanche, les argiles oxyhydroxydes de même que les silicates de type 1:1, en devenant porteurs de charges positives dans des conditions acides, offrent une grande capacité d’échange d’anions.
Tableau 5.3. CEC de quelques colloïdes du sol. Adapté de Brady and Weil (2010) et de Soil Survey Staff (2014)
| Colloïde | Étendue des valeurs de CEC [cmol(+) kg-1] |
|---|---|
| Kaolinite | 2 to 16 |
| Chlorite | 10 to 40 |
| Mica hydraté (illite) | 20 to 40 |
| Montmorillonite | 60 to 100 |
| Vermiculite | 100 to 150 |
| Matière organique (humus) | 150 to 400 |
| Aluminosilicates amorphes | 5 to 350 |
| Oxyhydroxyde de Fe/Al | ~0 to 3 |
ÉCHANGE DE CATIONS DANS LE SOL
Les surfaces des particules colloïdales du sol chargées négativement attirent les cations par les forces électrostatiques. Les particules colloïdales du sol qui retiennent les cations peuvent aussi les libérer en échange d’autres cations présents dans la solution de sol. Les cations les plus couramment retenus par les particules colloïdales du sol sont le Ca2+, Mg2+, K+, Na+, H+ et Al3+. Parmi eux, le Ca2+, Mg2+, K+, et Na+ sont des cations qui forment des bases, tandis que H+ and Al3+ e trouvent en plus grande abondance dans les sols acides.
L’échange de cations est le processus par lequel les cations de la solution du sol prennent la place des cations attachés à la surface des minéraux argileux (colloïdes inorganiques) et de l’humus (colloïdes organiques). L’échange de cations consiste en une permutation de place : un cation s’approprie la place d’un autre sur la surface d’un colloïde, tel que l’illustre la Figure 5.7 ci-dessous.

Les réactions d’échange de cations sont dites stœchiométriques, c’est-à-dire que les réactifs et les produits de la réaction demeurent dans les mêmes proportions. Ainsi, dans le cas des échanges divalents-monovalents (p. ex. entre Ca2+ et K+), un cation divalent échange sa place avec deux cations monovalents. Cette réaction presque instantanée atteint rapidement l’équilibre. Habituellement, la réaction d’échange de cations est réversible, à moins que le cation n’ait plutôt pris place dans une intercouche de phyllosilicate (p. ex. K+ dans du mica/illite hydraté).
La capacité d’échange cationique (CEC) est définie comme étant la quantité totale de cations échangeables qu’un sol peut adsorber (Brady et Weil, 2010). La CEC est généralement exprimée en centimoles de charge positive par kilogramme de sol (cmol(+) kg-1) ou en millimoles de charge positive par kilogramme de sol (mmol(+) kg-1). Ainsi, un sol caractérisé par une CEC de 10 cmol(+) kg-1 peut adsorber 10 cmol d’un cation monovalent (p. ex. Na+) ou 5 cmol d’un cation divalent (p. ex. Ca2+). Dans cet exemple, si ces 10 cmol de Na+ avaient pris la place d’un cation monovalent dans le processus d’échange (p. ex. K+), ce sont 10 cmol de K+ qui auraient cédé leur place. Cependant, si ces 10 cmol de Na+ avaient pris la place d’un cation divalent dans le processus d’échange (p. ex. Ca2+), ce sont 5 cmol de Ca2+ qui auraient cédé leur place.
Étant donné que ce sont les charges négatives (y compris les charges négatives influencées par le pH) présentes à la surface des colloïdes du sol qui sont en cause dans le processus de CEC, la nature et la quantité de colloïdes inorganiques (type d’argiles en présence) et de colloïdes organiques, de même que le pH du sol sont les principaux facteurs de détermination de la CEC d’un sol.
Matière à réflexion
Avec la photosynthése et la respiration, aucun processus dans la nature n’est probablement aussi vital pour la vie végétale et animale que l’échange d’ions entre les particules du sol et les racines des plantes.
Nyle C. Brady
Détermination de la Capacité d’Échange Cationique
On détermine généralement la CEC en procédant par étape : (a) saturation de tous les sites d’échange avec un cation de référence ; élimination des cations de référence en excès (lessivage 1) (b) remplacement de ce cation de référence avec un autre cation (lessivage 2) (c) mesure du cation de référence qui se trouve ainsi libéré dans la solution (lessivat). L’étape de saturation consiste à saturer avec un cation de référence (p. ex. Na+ ou NH4+) une masse connue de sol séché au four. Pour ce faire, on prépare une solution (généralement de l’acétate de sodium ou d’acétate d’ammonium) contenant le cation de référence tamponnée à pH 7.0. C’est avec cette solution que l’on va saturer tous les sites d’échange. Cette étape comprend aussi l’élimitation dans l’échantillon de sol de tout excès de cations de référence par lessivage avec une solution non polaire, telle que l’éthanol à 95 %. Il faudra bien sûr vérifier au préalable que tous les cations sur les sites d’échange ont bien été tous remplacés par un cation de référence. La deuxième étape comprend aussi un lessivage. Il consistera à déloger tous les cations de référence retenus sur les sites d’échange avec une solution contenant un cation différent (p. ex. K+). La concentration de cations de référence retrouvée dans le lessivat servira de mesure au calcul de la CEC du sol, dernière étape de la détermination de la CEC (Hendershot et coll., 1993).
Saturation en Bases
La proportion de CEC saturée par les cations à l’origine de la formation des bases (généralement Ca2+, Mg2+, K+, et Na+) est connue sous le nom de « saturation en bases ». Le pourcentage de saturation en bases est calculé par le recours à l’équation :
(1) 
La CEC et la somme des cations échangeables formant une base doivent être exprimées dans les mêmes unités (p. ex. en cmol(+) kg-1). Étant donné que les cations échangeables prédominants formant une base dans le sol sont le Ca2+, le Mg2+, le K+, et le Na+, l’équation se présente le plus souvent de cette façon :
(2) 
On notera que dans le cas des cations bivalents, tels que le Ca2+ et le Mg2+, les cations échangeables en cmol (+) doivent être calculés en fonction de la charge, soit 1 cmol de Ca2+ = 2 cmol de charge (+). La saturation en bases augmente avec le pH du sol. La saturation en bases est faible dans un sol à pH faible (< 7). Et, à mesure que le pH du sol augmente, la saturation en bases augmente aussi, et presque de façon linéaire entre pH 4 et pH 7.
ADSORPTION ET ÉCHANGE D’ANIONS
Certains colloïdes du sol, dont la surface se trouve chargée positivement dans des conditions de pH du sol < 7, sont capables de retenir des anions de façon similaire à la rétention des cations. On distingue deux mécanismes d’adsorption des anions : le mécanisme non spécifique régi par les forces électrostatiques des surfaces colloïdales chargées positivement et le mécanisme spécifique régi par les liens chimiques. Certains anions sont retenus par des mécanismes d’adsorption non spécifiques, d’autres, par des mécanismes d’adsorption spécifiques.
Dans le mécanisme d’adsorption non spécifique, les colloïdes dont la surface se trouve chargée positivement adsorbent les anions, mais les forces électrostatiques qui les retiennent sont faibles. Les anions les plus adsorbés selon ce mécanisme sont surtout le chlorure (Cl–) et le nitrate (NO3–). Du fait de leur faible retenue sur les surfaces chargées positivement, ces anions se retrouvent facilement remplacés par d’autres anions présents dans la solution de sol ; ils se trouvent tout aussi facilement évacués du sol par simple lessivage. Le mécanisme d’adsorption non spécifique n’a lieu sur les surfaces colloïdales que si elles sont chargées positivement. Or, on a dit que le pH < 7 favorisait l’ionisation positive des surfaces. On en déduit donc qu’un sol doté de minéraux dont la charge de surface colloïdale varie en fonction du pH aura une capacité d’échange d’anions élevée à bas pH. Ainsi en est-il des sols des tropiques qui contiennent des proportions élevées d’oxyhydroxydes et de kaolinite ; dans des conditions de bas pH, ces minéraux présentent des charges positives propices à l’adsorption non spécifique des anions.
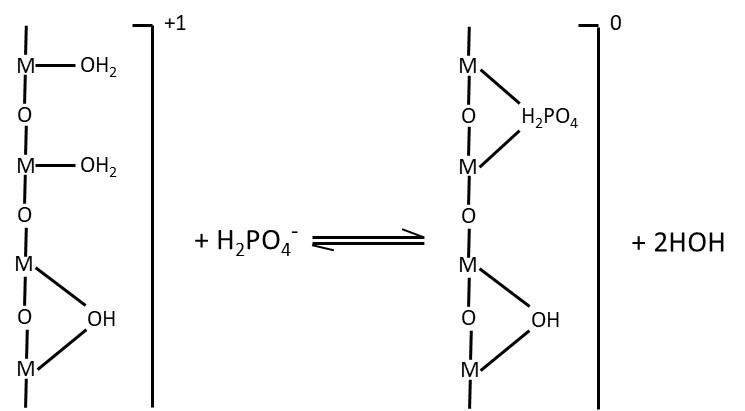
Dans le mécanisme d’adsorption spécifique, ce ne sont plus les forces électrostatiques qui retiennent les anions à la surface des colloïdes, mais ce sont des liens plus forts, de nature chimique. Le caractère spécifique du mécanisme d’adsorption porte sur l’anion adsorbé. Les anions phosphate (p, ex., H2PO4–) peuvent se retrouver adsorbés par échange spécifique entre un hydroxyle de surface contre un atome d’oxygène de l’anion phosphate (Figure 5.8). Par la force du lien qui le retient au colloïde, il est très peu probable que l’anion phosphate soit libéré de nouveau dans la solution de sol ; aucune plante ne pourra alors l’absorber. Il arrive même que toute la surface d’origine des colloïdes de ces minéraux se trouve couverte de l’ion adsorbé. Les anions les plus communs qui sont retenus par adsorption spécifique sur les colloïdes de ces minéraux comptent les anions phosphate, sulfate, silicate et carbonate.
pH DU SOL
Le pH du sol est une mesure de l’activité (concentration) des ions hydrogène dans la solution de sol ; elle définit son caractère acide ou alcalin. Dans ce contexte-ci, il ne sera pas nécessaire de faire la distinction entre « activité » et « concentration », comme le font les chimistes. Les deux termes définissent un rapport de quantité d’ions présents dans un volume de solution donné. La distinction réside dans la diversité d’ions présents dans la solution ; plus elle est grande, moins chaque ion est actif au sens chimique du terme. Le terme « activité » s’emploie pour désigner la capacité maximum d’agir d’un ion présent dans une solution. Dans cette situation, l’activité de l’ion donné égale celui de sa concentration, ce qui n’est pas le cas si la solution contient beaucoup d’autres ions (désigné par l’appellation « concentration »). Or, comme la solution dont il est question ici est la solution de sol, considéré comme très peu abondante autant en termes de diversité que de quantité, on assume que les deux termes expriment le même rapport.
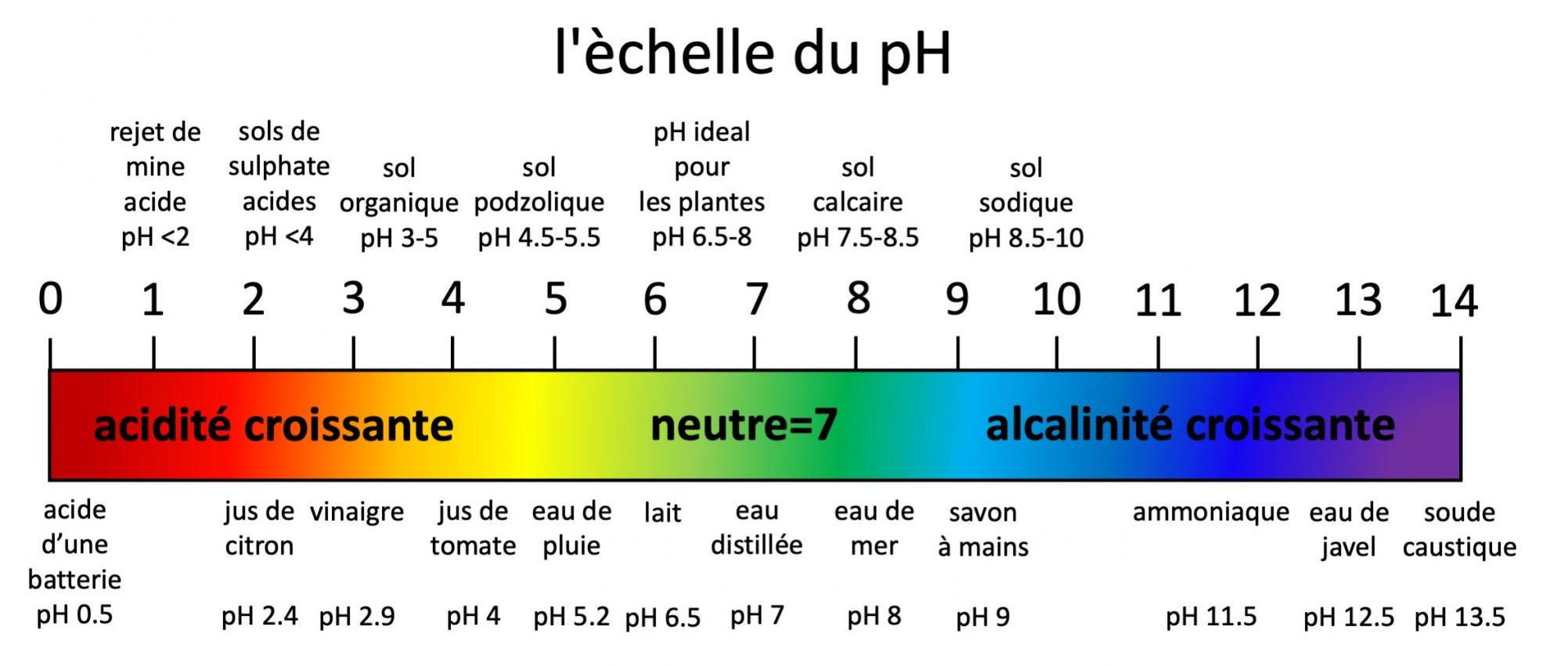
On présente les valeurs de pH sur une échelle standard qui varie de 0 à 14. Le pH 7 exprime la concentration des ions d’hydrogène d’une solution neutre, donc ni acide ni basique. Le pH de l’eau pure est de 7. Les valeurs de pH inférieures à 7 correspondent à l’acidité, qui vont croissantes jusqu’à la valeur maximale de 0. Les valeurs de pH supérieures à 7 correspondent à l’alcalinité qui vont croissantes jusqu’à la valeur maximale de 14. L’échelle de valeurs du pH est logarithmique, ce qui signifie, par exemple, qu’un sol de pH 4 a 10 fois plus d’ions hydrogène en solution qu’un sol de pH 5 et 100 fois plus qu’un sol de pH 6 et ainsi de suite. Par exemple, un sol à pH 6 a dix fois plus d’ions hydrogène en solution qu’un sol à pH 7, et un sol à pH 8 a dix fois moins d’ions hydrogène qu’un sol à pH 7. La Figure 5.9 présente l’échelle standard du pH ainsi que quelques exemples de pH de sols et de produits communs.
Les principaux facteurs qui influencent le pH naturel du sol comptent le matériel géologique dont il tire son origine et les produits de sa propre réaction au dioxyde de carbone (CO2) dissous qu’apporte l’eau des précipitations. Un autre facteur d’influence du pH du sol réside aussi dans la présence d’acides organiques résultant de la décomposition de la végétation. Au Canada, la très grande majorité des sols minéraux se sont formés à partir de deux types de matériau parental : l’un granitique acide, l’autre, carbonaté alcalin. Le matériau parental granitique dérive principalement des roches du Bouclier précambrien, tandis que le matériau parental carbonaté dérive principalement des roches de l’ère paléozoïque et de roches plus récentes. Les sols formés à partir du matériau parental granitique présentent l’étendue des valeurs de pH caractéristiques des sols acides. En revanche, les sols formés à partir de matériau parental carbonaté présentent l’étendue des valeurs de pH caractéristiques des sols neutres ou alcalins.
Le pH de l’eau de pluie gravite autour de 5,6, une valeur qui découle de la réaction de l’eau avec les gaz de l’atmosphère, particulièrement le CO2. Cependant, son pH va baisser s’il y a présence de polluants atmosphériques (p. ex. le SO2 forme de l’H2SO4 en s’hydrolysant). De manière générale, les sols tendent tous à s’acidifier avec la durée d’exposition aux précipitations, laquelle peut mener au lessivage des cations basiques. Étant donné que le matériau parental granitique contient très peu de matière minérale basique, les sols qui en dérivent ont tendance à s’acidifier plus facilement que ceux qui dérivent du matériau parental carbonaté. Les sols podzoliques, généralement développés sur du matériau parental granitique, sont caractérisés par des pH qui varient entre 4,5 et 6,0. Les sols carbonatés, généralement développés sur du matériau parental calcaire, se trouvent à neutraliser les effets acidifiants du CO2 dissous apporté par les précipitations. Ces sols sont caractérisés par des pH dont les valeurs se situent entre 7,5 et 8,5.
Certains sols sont en mesure de tamponner le pH de la solution de sol. En effet, bien que la valeur d’une mesure de pH ne représente qu’une concentration ponctuelle de H+ en solution, cette valeur demeure relativement stable, car des sites d’échange appelés « acidité échangeable » adsorbent des H+ qui, grâce à leur pouvoir tampon, stabilisent le pH en solution. Or, les sols à CEC élevée témoignent de la présence élevée de ces sites d’acidité échangeable. Grâce à leur capacité de maintenir le pH du sol stable en échangeant des H+ avec la solution de sol, l’ajout de composés acides ou basiques (p. ex. un engrais) ne provoquera pas de changement de pH. On distingue deux types d’acidité : l’acidité active, révélée dans la mesure ponctuelle du pH de la solution de sol et l’activité d’échange, celle présente sur les sites d’échange. Les sols à forte teneur en argile ou en matière organique bénéficient d’un grand pouvoir tampon, parce qu’ils sont dotés d’une grande réserve d’acidité d’échange, ce qui n’est pas le cas des sols sableux à faible teneur en matière organique.
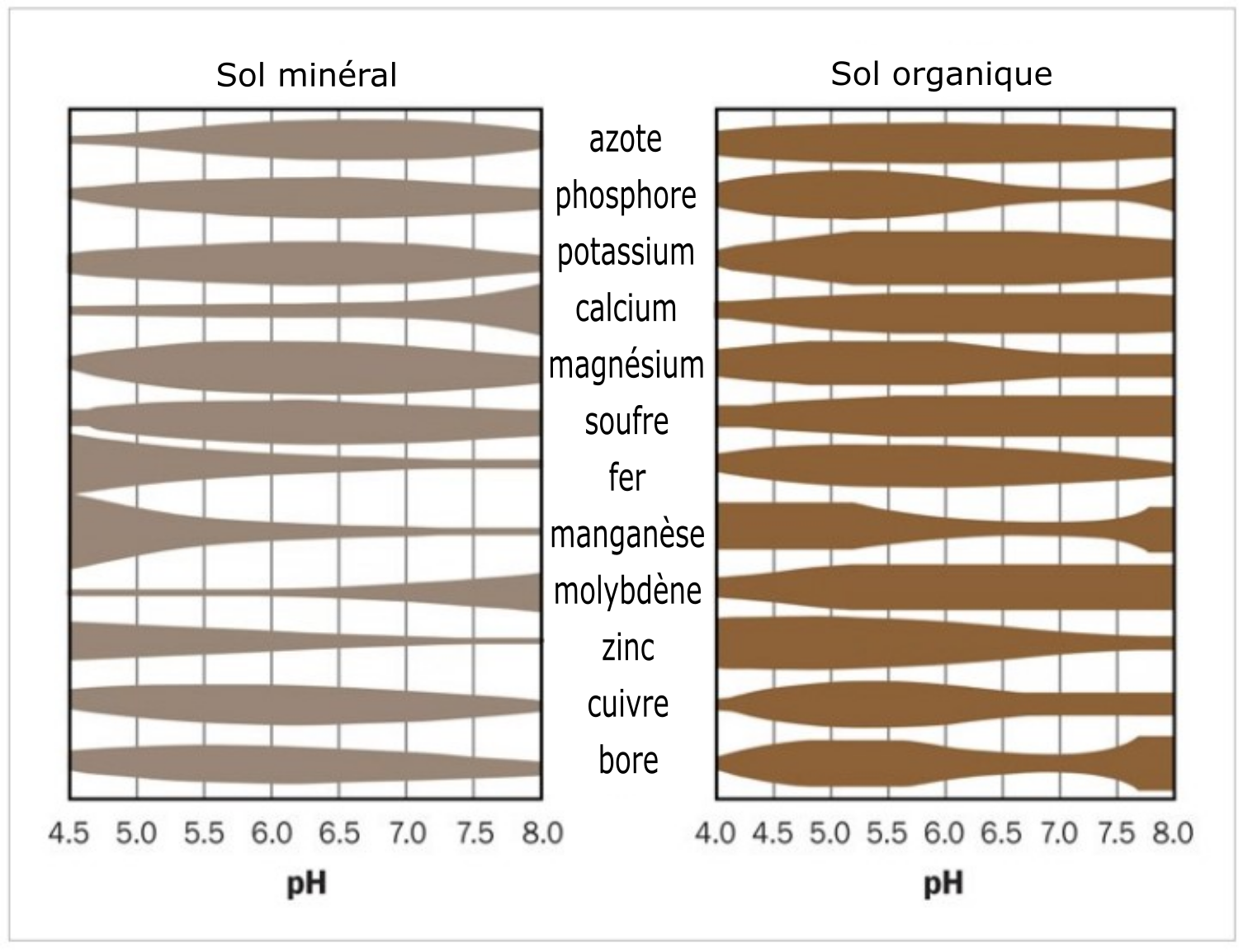
Le pH du sol agit sur la disponibilité des éléments nutritifs nécessaires aux plantes. Tous les éléments nutritifs ne présentent pas la même disponibilité à un pH donné. La disponibilité des éléments nutritifs pour les plantes est optimale à des valeurs de pH qui varient entre 6,5 et 8,0. Les conditions de pH extrêmes entravent l’absorption des éléments nutritifs pour les plantes et peuvent créer des effets nocifs sur elles. Par exemple, une solution de sol très acide a pour effet d’augmenter la solubilité de l’aluminium, du manganèse et du fer qui, à des concentrations élevées, deviennent potentiellement toxiques pour les racines des plantes. À des pH très acides, ces éléments peuvent réagir avec les éléments nutritifs nécessaires aux plantes, avec pour effet de les rendre indisponibles pour elles. À des pH très alcalins, soit à l’autre extrême de l’échelle de valeurs de pH, du Ca2+ peut être libéré en excès dans la solution de sol, le faisant réagir avec d’autres éléments nutritifs (p. ex. le phosphate), les rendant tout aussi indisponibles pour les plantes. La Figure 5.10 schématise la disponibilité des éléments nutritifs des plantes dans les sols minéraux et organiques en fonction des valeurs de pH.
Acides et Bases
Les différentes valeurs de pH témoignent de la diversité des réactions qui mettent en jeu le couple acide-base dans l’eau, qui se dissocie en :
H2O ↔ H+ + OH–
Une solution aqueuse est dite neutre à pH 7 ; la concentration en protons [H+] se trouve alors exactement égale à la concentration en ions hydroxyles ([OH–]), les deux étant égaux à 10-7 moles par litre.
Un acide définit tout composé chimique dissout dans l’eau capable de libérer un proton, tandis qu’une base définit tout composé capable d’accepter un proton. Lorsqu’un acide perd un proton, il forme la base conjuguée (on dit aussi « partenaire basique ») :
HL ↔ H+ + L–
Dans l’équation ci-dessus, HL représente l’acide capable de donner un proton (H+) en solution et L– ireprésente la base conjuguée, capable d’accepter un proton. Dans cette équation, la base conjuguée (L–) est également appelée « ligand » (on dit aussi « coordinat »). La force d’un acide est donnée par sa capacité à se dissocier dans l’eau et à libérer des protons en solution. L’acide sulfurique, par exemple, est un acide fort, car en solution tous ses protons et ligands se dissocient.
Une base est un composé chimique capable de générer des ions hydroxyles (OH–) ou d’absorber des ions H+ en solution pour former son acide conjugué (on dit aussi « partenaire acide »).
B + H2O ↔ OH– + BH+
La matière organique humifiée du sol constitue une source de groupes fonctionnels de surface qui influencent la chimie du sol, y compris le pH. Les groupes fonctionnels organiques les plus importants comprennent les groupes carboxylique (-COOH), phénolique-OH, alcoolique-OH, carbonyle (C = O) et quinone, ainsi que les groupes amine (-NH2) et thiol (-SH). Les groupes fonctionnels acide carboxylique, phénolique-OH et alcool-OH contribuent à créer l’acidité et les échanges d’ions. Ces groupes, qui ne se dissocient pas complètement en solution, sont qualifiés d’acides faibles. Ils constituent l’acidité d’échange présente sur les sites d’échange de cations. D’autres groupes fonctionnels, tels que les acides aromatiques, présentent peu de dissociation en solution et jouent sur les sites d’échange le même rôle de réserve d’acidité et d’échange de cations.
Le pH du sol gouverne bon nombre des processus chimiques qui ont lieu dans le sol, en particulier celui qui rend les éléments nutritifs disponibles aux plantes (Ca2+, Mg2+, K+, etc.). La plupart des sols ont un pH qui varie entre 5 et 8 ; le pH optimal pour la croissance de la plupart des plantes cultivées en sol canadien se situe entre 5,5 et 7,5. Mais de nombreuses espèces ont pu s’adapter à des valeurs de pH hors de ces limites. L’exemple des bleuets est bien connu. Les Vaccinium, genre auquel l’arbuste aux petits fruits bleus appartient, croissent mieux dans un sol acide (pH variant entre 4,3 et 5,5) que dans un sol alcalin (pH > 7).
Les milieux alcalins peuvent influencer la disponibilité des éléments nutritifs pour les plantes. Par exemple, les sols comprenant des concentrations naturellement élevées de Na+ ou ceux qui ont reçu de grandes quantités de Na2CO3 (bicarbonate de sodium) par irrigation peuvent avoir des pH de 8,5 ou plus. Ces valeurs de pH résultent de la présence de précipités de Na2CO3 dans le sol. Or, si un sol contient beaucoup plus d’ions Na+ que d’ions Ca2+ ou d’ions Mg2+ dans la solution du sol, il risque d’en accumuler, une situation qui peut être préoccupante dans les régions arides ou semi-arides, telles que les Prairies canadiennes, où se trouvent les sols solonetziques connus pour leur haute teneur en Na.
Mesure du pH du Sol
Le pH est sans aucun doute la propriété chimique du sol la plus mesurée. On mesure généralement le pH du sol à l’aide d’un pH-mètre. L’appareil mesure la différence de potentiel existant entre une électrode de référence et une autre électrode de mesure, toutes deux plongées dans la solution dont on veut connaître le pH. L’utilisation que l’on souhaite faire des mesures de pH détermine la façon de procéder pour les obtenir. Les procédés de mesures font intervenir les paramètres suivants : 1) rapport sol / solution, 2) l’ajout ou non d’un électrolyte, 3) la durée de repos de l’échantillon de sol avant la prise de mesure. Les procédés comprennent, mais sans s’y limiter : rapport sol : eau 1:1, 1:2, 1:2.5, 1:5 et 1:10 (appelé aussi « pâte saturée ») ; rapport sol : 0,01 M CaCl2 ou 1M KCl ou 1.0M NaF (Soil Survey Staff, 2014; Hendershot et coll., 2008). Ce qu’on mesure est bien le pH de la solution de sol et non des particules de sol elles-mêmes (Hendershot et coll., 2008). Les mesures de pH du sol effectuées dans des suspensions d’électrolyte (p. ex. rapport sol:solution de 0.01 M CaCl2 1:2) sont généralement plus stables et reproductibles que celles effectuées dans H2O, en raison de la constance de la concentration de l’électrolyte. Cependant, l’ajout d’un électrolyte a généralement pour effet de faire diminuer le pH du sol par rapport au seul ajout d’eau. Par exemple, les valeurs mesurées dans le rapport sol:eau sont habituellement de 0,5 à 0,6 unité plus élevées que celles mesurées dans le rapport sol:solution 0.01M CaCl2, et de 0,7 unité plus élevées que celles mesurées dans le rapport sol:solution 1M KCl. Les cations (Ca2+ ou autres) — provenant de l’électrolyte — passent en solution avant d’être adsorbés sur les sites d’échange, ces derniers libérant, par effet de remplacement, des cations acides en réserve, tels que Al3+ ou H+, d’où l’augmentation d’acidité.
Dans un sol contenant un minéral carbonaté, on obtient généralement des valeurs de pH qui varient entre 7,6 et 8,5. Cette variation de mesures est attribuable à quelques facteurs clés. Premier facteur : libération d’Al3+ et de Fe3+ dans la solution de sol. En plus des ions échangeables sur les sites d’échange des colloïdes, tels que le Ca2+, le Mg2+, le Na+ et le K+, de petites quantités d’Al et de Fe échangeables présents dans le colloïde s’hydrolysent lorsqu’ils sont libérés dans la solution de sol, entraînant une diminution du pH. Deuxième facteur : lenteur de réaction de la dolomite minérale [Ca,Mg(CO3)2]. Étant donné que la dolomite minérale réagit (se dissout) beaucoup plus lentement que la calcite (CaCO3), on doit respecter le laps de temps nécessaire à son temps de réaction si l’on souhaite obtenir la juste valeur du pH (p. ex. 60 min). Troisième facteur : le non-respect des recommandations à l’égard du temps d’attente. Il est d’usage d’attendre au moins une heure avant de prendre une mesure de pH, le temps que les ions en suspension dans l’échantillon se stabilisent (voir Hendershot et coll., 2008). Une telle recommandation de temps standardisé se révèle utile aux grands laboratoires commerciaux d’analyses de sols qui régissent leurs opérations suivant la norme internationale ISO17025. Bien que ce laps de temps suffise pour que les échanges d’ions avec la solution s’équilibrent, il peut toutefois se révéler insuffisant pour l’atteinte de l’équilibre entre les échanges de carbonates (de dissolution lente) et le CO2 de l’atmosphère. D’autres facteurs peuvent également exercer une influence sur la mesure du pH.
Adjustement du pH du Sol
Le sol montre une tendance naturelle à s’acidifier sous l’influence de l’eau de pluie, de la décomposition de la matière organique et des applications d’engrais, tels que l’urée et l’ammoniac anhydre. L’ajout de pierre à chaux agricole peut faire augmenter le pH du sol pour le besoin des cultures. La quantité de chaux agricole à appliquer et le meilleur moment de l’appliquer dépendent de la culture que l’on veut produire. Il est généralement conseillé de recourir à de la chaux lorsque le pH du sol passe en dessous de 6,0 (Munro, 2018). De la pierre calcaire finement broyée (principalement composée de calcite ; CaCO3) ou de la dolomie (principalement composée de dolomite ; Ca,Mg(CO3)2) sont les produits de chaulage les plus fréquemment utilisés, bien que de nombreux autres produits ayant une capacité de neutralisation de l’acidité puissent l’être aussi.
Matière à réflexion !

Le programme de reverdissement de Sudbury
Dans la région de Sudbury, l’effet domino d’un siècle d’exploitation d’une fonderie de métaux a été impressionnant : rejets polluants de dioxyde de soufre, de cuivre, de nickel sont devenus cause initiale d’accroissement des incendies, d’érosion des sols et de gels sur des milliers d’hectares de terres qui s’étaient si érodées et détériorées avec le temps qu’à peine quelques petits boisés d’érables et de bouleaux rabougris ont réussi à survivre. Aucune plante ne semblait désormais capable de recoloniser ces sols podzoliques à texture grossière, déjà peu résistants à la sécheresse et pauvres en éléments nutritifs, qui s’étaient en plus érodés et acidifiés avec les années. La perte graduelle de la végétation avait fini par priver les sols de toute protection, ce qui avait fait disparaître l’humus, qui se trouvait désormais exposé aux effets du gel, du vent et de l’eau (en excès ou en déficit). Les rejets polluants les avaient rendus toxiques, notamment en raison de la présence d’aluminium soluble, impropres à l’établissement de semis. Des résultats de recherches sur le terrain dans les années 1970 ont permis de démontrer l’effet positif de l’application en surface de calcaire dolomitique moulu [principalement CaMg(CO3)2] au taux de 3 tonnes à l’hectare, les valeurs de pH du sol ayant remonté – de moins de 3,0 à entre 5,2 et 5,6. De plus, avec la mise sur pied d’un programme de détoxification des sols destiné à favoriser l’installation de la végétation, on a combiné traitement par chaulage (avec ou sans un engrais) et ensemencement de graminées-légumineuses. Des espèces ligneuses telles que le bouleau, le tremble et les saules ont repoussé naturellement. Dans le but d’aider à poursuivre la colonisation, on a planté près de 10 millions d’arbres dans la région de Sudbury entre 1976 et 2020, un mélange de conifères et de feuillus indigènes destinés à produire la source de graines nécessaire à cette fin.
D’autres cultures nécessitent des conditions de sol plus acides, telles que la culture des pommes de terre, des bleuets, d’azalées et certaines autres cultures horticoles. Il est toutefois plutôt rare que l’on doive créer des conditions plus acides par ajustement intentionnel du pH. Cela se fait néanmoins à l’aide d’ajout de soufre élémentaire ou d’engrais contenant de l’ammoniac.
SALINITÉ ET SODICITÉ DU SOL
La teneur en sels dans un sol, soit la salinité, est régie par la concentration de la totalité des sels minéraux dissous dans la solution du sol. La présence de sels dans le sol et dans l’eau est tout à fait naturelle. Mais il arrive que dans certaines conditions, le sol puisse en accumuler. L’accumulation de sels solubles dans le sol définit le phénomène de salinisation. Les sels dissous qui contribuent à la salinité du sol comprennent les cations sodium (Na+), calcium (Ca2+, Mg2+), et potassium K+; et les anions chlorure (Cl−), sulpfate (SO42−), bicarbonate (HCO3−), carbonate (CO32−), et nitrate (NO3−).
La sodicité est liée à la salinité, mais porte particulièrement sur le rapport entre la concentration de Na+ dans la solution de sol et les concentrations respectives de Ca2+ et Mg2+. Le degré de sodicité, estimé par le calcul du rapport d’adsorption du sodium (RAS), est une mesure du rapport de la concentration de Na+ sur les concentrations de Ca2+ et de Mg2+ dans l’extrait de sol saturé. Le RAS résulte du calcul de la concentration Na+ divisé par la racine carrée des concentrations moyennes [(Ca2+ + Mg2+)/2]. On utilise la racine carrée des concentrations [Ca2+ + Mg2+], car il s’agit d’ions divalents, tandis que Na+ est monovalent. On évalue la sodicité en pourcentage de sodium échangeable (PSE), autrement dit de Na+ échangeable exprimé en pourcentage de capacité d’échange de cations. Si le RAS du sol égale ou excède 13 ou si le PSE du sol égale ou excède 15 %, le sol est dit sodique.
(3) ![\begin{equation*}RAS = \frac{[Na^{+}]\;(mmol\;L^{-1})}{(0.5\times[Ca^{2+} + Mg^{2+}])^{\frac{1}{2}}\;(mmol L^{-1})}\end{equation*}](https://openpress.usask.ca/app/uploads/quicklatex/quicklatex.com-65b9f7eade132bd06f683f7879c0b768_l3.svg "Rendered by QuickLaTeX.com")
(4) ![\begin{equation*}PSE = \frac{[Na^{+}]\;(cmol(+)\;kg^{-1})}{CEC\;(cmol(+) kg^{-1})} \times 100\end{equation*}](https://openpress.usask.ca/app/uploads/quicklatex/quicklatex.com-292536f1f9d7ca4ec3296e765e22e860_l3.svg "Rendered by QuickLaTeX.com")
Salinité Progressive dans les Sols
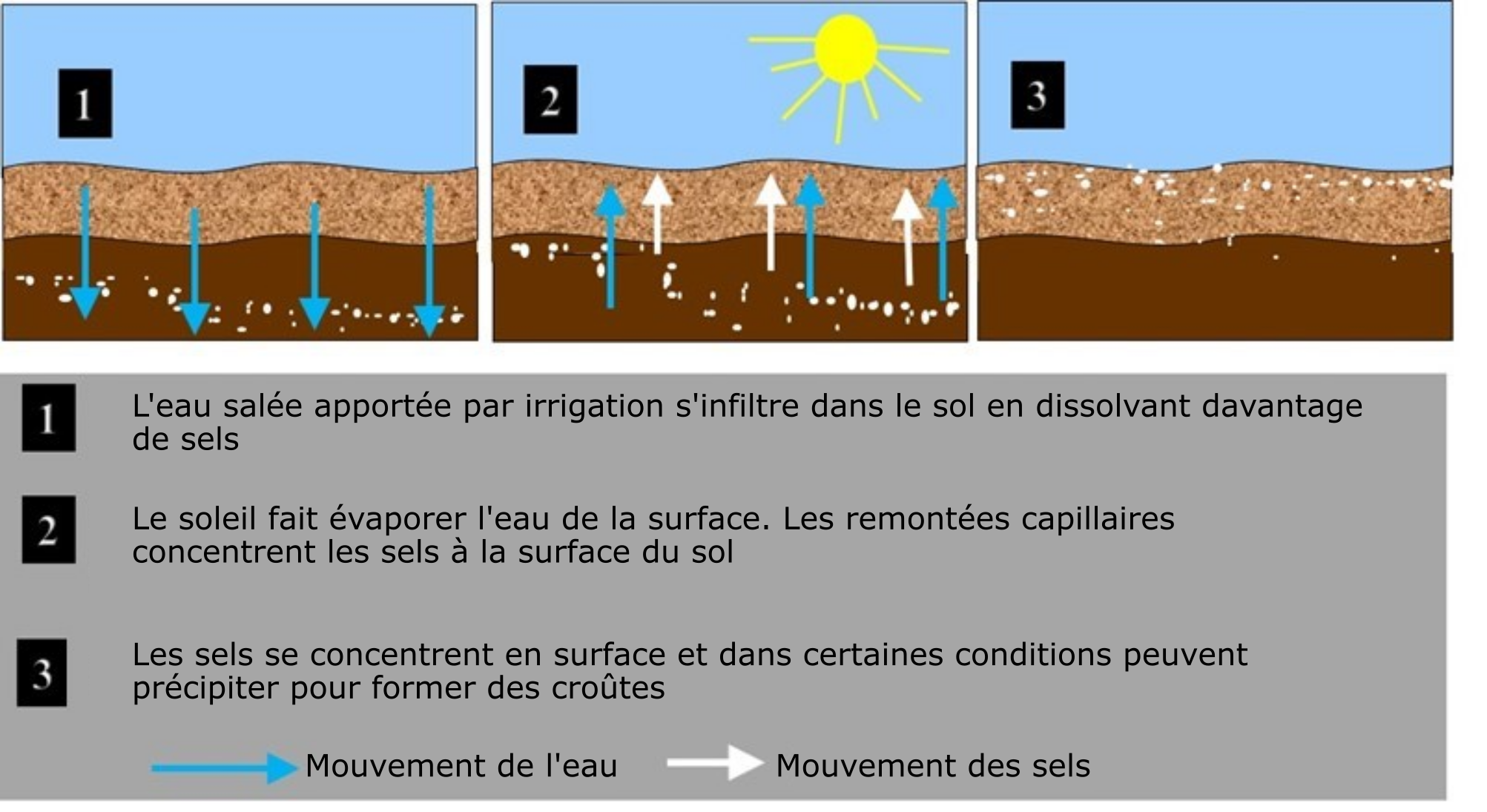
Dans un sol, les sels proviennent de deux sources : les naturelles et les anthropiques. Les sels de source naturelle proviennent de l’altération des minéraux primaires, des dépôts atmosphériques, des intrusions d’eau de mer, des rejets d’eaux souterraines salines et des saumures des gisements naturels de sel. Les sels de source anthropique proviennent de l’eau d’irrigation à fortes teneurs en sels (figure 5.11) et des engrais synthétiques et organiques à fortes teneurs en sels, tels que le fumier, les biosolides et le compost. Les sels de voirie visant à faire fondre la glace sur les routes pendant la saison hivernale au Canada constituent aussi une source anthropique de salinisation, bien qu’elle demeure localisée.
Divers facteurs contribuent à l’accumulation des sels solubles dans le sol :
- Conditions climatiques sèches – Deux facteurs d’accumulation de sels solubles dans le sol ont pour origine ces conditions. 1. Le peu de précipitations que reçoivent les sols dans les zones arides et semi-arides contribue à l’accumulation des sels solubles dans le profil, faute de lessivage suffisant. 2. L’évaporation élevée. Le mouvement ascendant de l’eau des couches profondes du sol vers la surface créé par évaporation élevée entraîne avec lui à la surface les sels solubles, qui forment des croûtes une fois l’eau évaporée. Dans les régions arides, on estime que le pourcentage de perte d’eau par évaporation élevée peut varier de 50 à 90 %, ce qui a pour effet de concentrer de 2 à 20 fois les sels solubles.
- Mauvais drainage – Un mauvais drainage (attribuable à une nappe phréatique élevée ou à une faible perméabilité du sol en profondeur) peut contribuer à faire augmenter la teneur en sels solubles du sol.
- Mauvaise qualité de l’eau d’irrigation – Les problèmes de salinité sont courants dans les terres irriguées, parce que l’eau d’irrigation contient souvent des concentrations très élevées de sels solubles. Bien souvent, les cultures n’utilisent qu’une partie de l’eau d’irrigation, le reste se trouvant à s’évaporer directement du sol humide, laissant à la surface les ions dissous, que l’on retrouve après sous forme de croûtes salines.
- Régions côtières, un facteur géographique – Les sols des zones côtières sont vulnérables à l’accumulation des sels provenant des intrusions d’eau de mer et des eaux souterraines salines.
- Autres facteurs d’accumulation de sels de source anthropique – Diverses autres pratiques de gestion contribuent à la salinisation des sols cultivés : surutilisation d’engrais synthétiques, application continue de fumier, apport des sels de voirie qui se retrouvent dans les fossés à proximité des champs cultivés ou directement sur les cultures, résultat des activités de déneigement.
Mesure de la Salinité du Sol
On mesure la concentration totale de sels dissous en extrayant un échantillon de sol avec de l’eau et en mesurant les concentrations de chaque sel présent dans l’extrait. Cette méthode permet de mesurer avec précision la concentration de chaque sel. En revanche, elle se révèle gruge-temps, laborieuse et coûteuse. Toutefois, sachant qu’il est possible de mesurer la conductivité électrique (CE) d’une solution et que cette dernière augmente proportionnellement avec la concentration totale de sels dissous, la CE peut servir d’indicateur de la salinité du sol. La mesure de la CE d’un extrait de sol est d’ailleurs la mesure à laquelle on recourt le plus souvent pour évaluer et définir la salinité totale d’un sol. La mesure de la CE – réalisée avec un conductimètre dans des laboratoires d’analyses de sols est obtenue rapidement – a l’avantage d’être peu coûteuse en plus de représenter une mesure fiable de la salinité. On détermine la CE d’un sol dans un extrait saturé (c.-à-d. dans la solution de sol extraite d’une pâte de sol saturée) ; les unités de mesure sont exprimées en déciSiemens par mètre (dS m-1) ou en milliSiemens par centimètre (mS cm-1). On retiendra que les sols dont la valeur de CE d’un extrait de sol saturé >4 dS m-1 entrent dans la classe des sols dits salins.
Propriétés des Sols Halomorphes
On distingue trois classes de sols halomorphes (sols touchés par la salinisation) suivant la concentration totale de sels Na+ dans le sol : les salins, les salins-sodiques et les sodiques. Les sols salins ont une concentration élevée en sels, mais leur concentration en Na+ est faible. Les sols sodiques ont une concentration relativement faible en sels, mais leur concentration en Na+ est élevée. Les sols salins-sodiques ont une concentration élevée et en sels et en Na+. La Figure 5.12 présente la classification des sols en fonction de la CE, du RAS et du PSE.
Les sols salins sont caractérisés par des concentrations élevées de sels dissous, dont les valeurs de CE sont supérieures à 4 dS m-1 (Figure 5.12). Leur concentration en Na+ dans la solution de sol ou sur le complexe d’échange est relativement faible, comme le témoignent les valeurs du RAS inférieures à 13 et les valeurs du PSE inférieures à 15 %. Le pH des sols salins est généralement inférieur à 8,5. Les concentrations élevées en sels caractéristiques de ces sols favorisent le phénomène de floculation des colloïdes du sol (attraction des colloïdes les uns envers les autres menant à la formation de flocs ou grappes).
Les sols sodiques sont caractérisés par de faibles concentrations de sels dissous, dont les valeurs de CE sont inférieures à 4 dS m-1. Leur concentration en Na+ dans la solution de sol ou sur le complexe d’échange est relativement élevée par rapport aux concentrations respectives de Ca2+ et de Mg2+. Par conséquent, les valeurs du RAS sont supérieures à 13 et les valeurs du PSE sont supérieures à 15 % (Figure 5.12). Le pH des sols sodiques généralement supérieur à 8,5 s’explique par l’hydrolyse du carbonate de sodium. La concentration élevée de Na+ échangeable dans ces sols mène à des réactions avec les colloïdes qui les font se gonfler, source de leur dispersion. Les phénomènes de gonflement et de dispersion sont d’autant plus importants que le complexe d’échange du colloïde hébergera une part importante d’ions hydratés et monovalents, principalement le Na+. Les ions Na+ hydratés de la solution de sol qui s’immiscent dans les particules d’argile neutralisent les forces électrostatiques qui les lient entre elles. L’apport d’un supplément de molécules d’eau forcera les particules à se séparer, phénomène à l’origine de celui de dispersion des matériaux de la matrice du sol. Le phénomène de dispersion touche aussi la matière organique du sol dans des conditions de pH élevé. Les sols sodiques alcalins appartenant aux Solonetz noirs ont de ces matériaux humiques de couleur sombre qui couvrent leurs mottes dispersées.
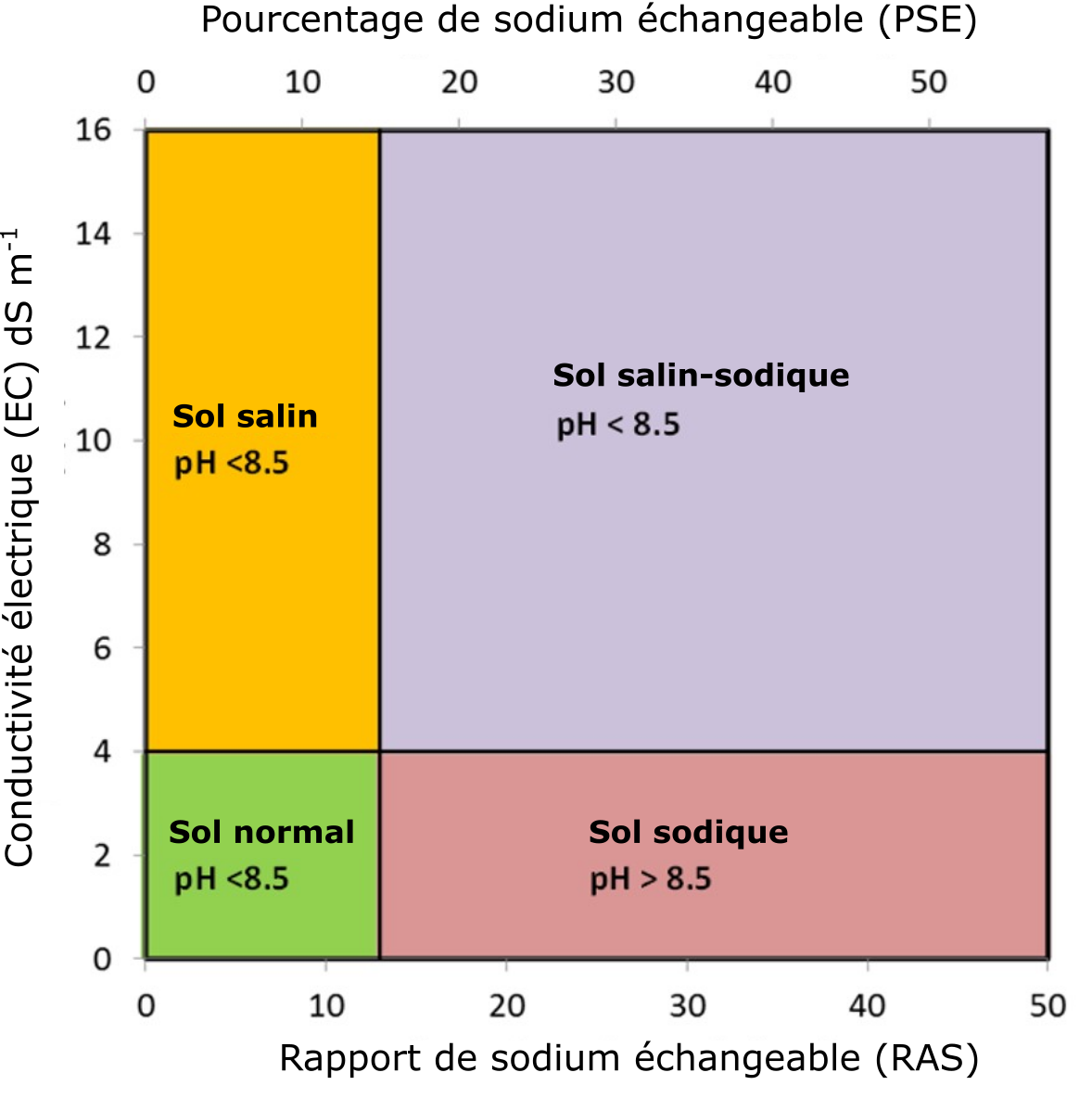
Les sols caractérisés par des concentrations élevées de sels dissous et de Na+ sont appelés sols « salins-sodiques ». Ces sols ont des valeurs de CE supérieures à 4 dS m-1, des valeurs de RAS supérieures à 13 et de PSE supérieures à 15 %. Le pH des sols salins-sodiques est généralement inférieur à 8,5. Le phénomène de floculation s’observe aussi dans ces sols, en raison de leur concentration élevée en sels dissous (Sparks, 2003).
Effets de la Salinité et de la Sodicité des Sols sur la Santé des Écosystèmes
La salinisation des sols, l’un des phénomènes les plus dévastateurs à l’échelle de la planète, est une cause sérieuse de dégradation des terres et de désertification des écosystèmes arides. Partout dans le monde, la salinisation des sols a contribué à réduire la superficie de terres cultivables et la production des cultures.
La salinité et la sodicité altèrent les propriétés du sol et compromettent la croissance des plantes et l’activité microbienne. Des concentrations élevées de sels dissous dans la solution de sol (soit un potentiel osmotique élevé, voir Chapter 4) réduisent la disponibilité de l’eau nécessaire à la croissance des plantes. En effet, l’absorption de l’eau par les racines des plantes passe par des mécanismes osmotiques qui sont régis par la présence de ces sels dissous dans la solution de sol. Les plantes ne présentent cependant pas toute la même sensibilité aux sols hautement salins. La sensibilité d’une plante à la salinité définit un état physiologique connue sous l’appellation « sécheresse physiologique », état qui rend les racines d’une plante incapables d’absorber l’eau, même s’il y en a suffisamment dans la solution de sol pour répondre à ses besoins. Les microorganismes du sol, qui absorbent l’eau suivant le même principe osmotique que les plantes, se heurtent aux mêmes difficultés d’absorption en présence de concentrations élevées de sels dissous dans la solution de sol.
Tableau 5.4. Sensibilité de certains végétaux aux conditions salines
| Sensible | Moyennement Sensible | Moyennement Tolérant | Tolérant |
|---|---|---|---|
| (CE < 4 dS m-1) | (CE 4-7 dS m-1) | (CE 7-9 dS m-1) | (CE 9-12 dS m-1) |
| légumes | luzerne | fétuque | orge |
| érable | maïs | peuplier | blé |
| tomates | graminées | avoine | saule |
| pomme de terre | trèfle | soya | |
| Note: Les plantes capables de survivre à valeurs de CE comprises entre 12 et 15 dS m-1 sont considérées comme halophytes ; cependant, certaines d’entre elles, voire très peu, peuvent survivre à des valeurs de >15 dS m-1 | |||
La forte concentration de Na dans les sols sodiques fait gonfler les minéraux argileux dotés de cette propriété de dispersion, cause d’affaiblissement de la structure du sol et de réduction de perméabilité et d’infiltration par blocage des pores du sol et de formation de croûtes de surface. Or, ce manque d’infiltration et de perméabilité peut aussi contribuer à l’érosion du sol, à accélérer la saturation du sol pendant l’irrigation, au cours d’événements de fortes pluies et de fonte de la neige, contribuant encore plus à boucher les pores du sol, canal d’entrée d’air indispensable à l’aération. Des conditions anaérobiques se créeront dans la foulée de tels événements, affectant négativement la croissance des plantes.
Sols Affectés par le Sel : Distribution dans le Monde et au Canada
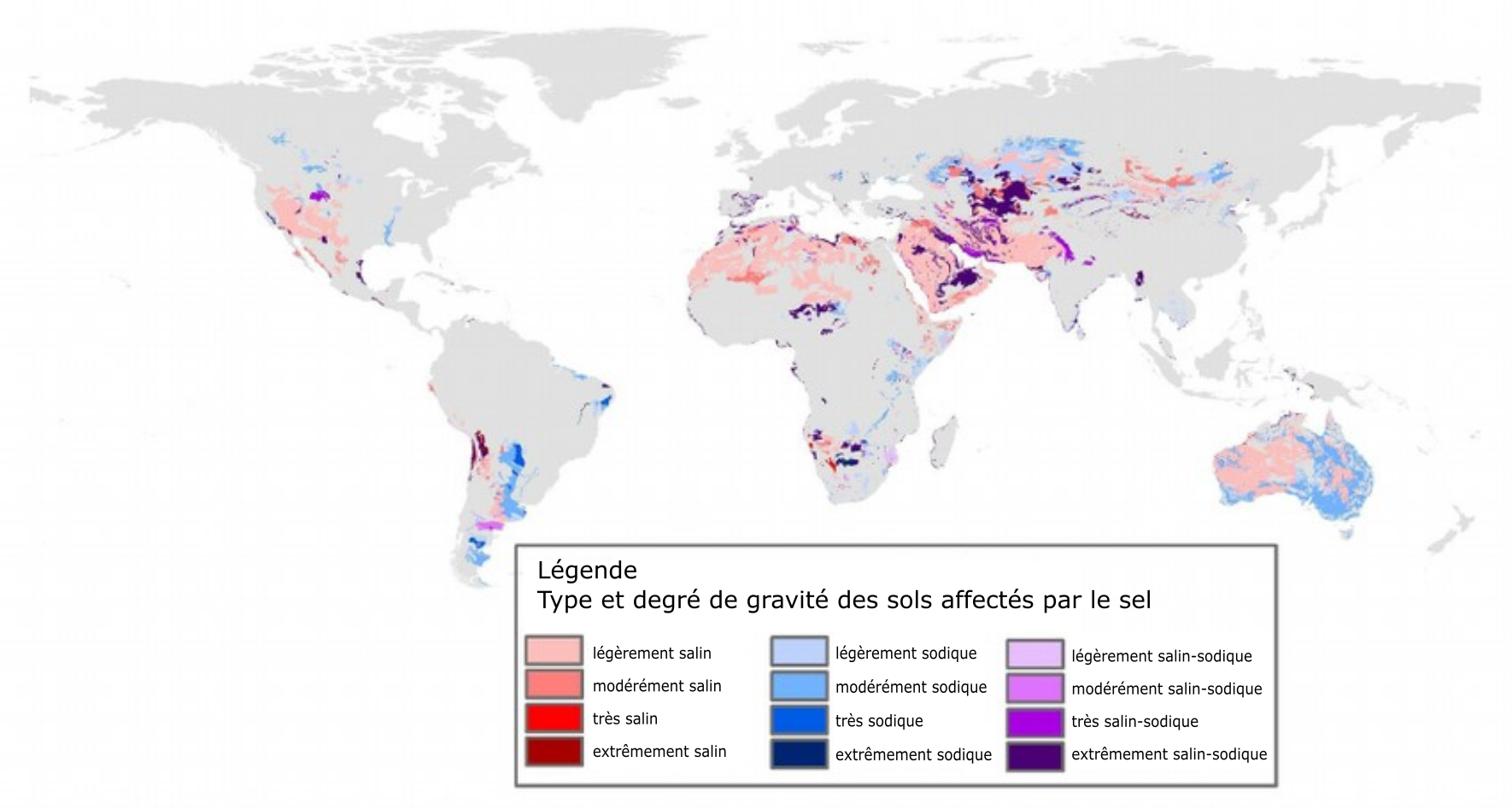
La salinisation des sols est un problème mondial, principalement dans les zones arides et semi-arides de tous les continents. La salinisation est moins préoccupante dans les zones humides, car les précipitations sont suffisantes pour lessiver du profil du sol les sels en excès. Les zones salinisées dans le monde augmentent à un taux d’environ 10 % par année, autant en raison de causes naturelles qu’anthropiques. Selon la cartographie mondiale des sols de 1970–1980, on a estimé à 397 millions d’hectares la superficie totale des sols salins et à 434 millions d’hectares celle des sols sodiques (FAO, 2020). La figure 5.13 montre la répartition des sols salins et sodiques dans le monde (Zheng, 2014). On a signalé le phénomène de dégradation des terres attribuable à la salinisation dans le bassin de la mer d’Aral (bassins des fleuves Amu-Darya et Syr-Darya), le bassin indogangétique en Inde, le bassin de l’Indus au Pakistan, le bassin du fleuve Jaune en Chine, le bassin de l’Euphrate en Syrie et en Irak, dans le bassin Murray-Darling en Australie et dans la vallée de San Joaquin aux États-Unis (Qadir et coll., 2014).
Au Canada, on estime qu’il y aurait entre 6 et 8 millions d’hectares de sols salins, que l’on trouve principalement dans les Prairies canadiennes (Miller et Brierley, 2011). En 1995, 34 % de la superficie des terres agricoles dans les provinces des Prairies (Acton et Gregorich, 1995) présentaient un risque de salinisation, qui allait de « modéré à très élevé » ; en 2001, le risque de salinisation ne concernait plus que 22 % de la superficie (soit une diminution de 12 %) (Eilers et coll., 2010). On attribue cette réduction de salinisation des terres agricoles aux bienfaits apportés par la réduction de la jachère d’été et de l’application de pratiques de conservation des sols, telles que l’établissement d’une couverture végétale permanente.
Matière à réflexion !
Salinité des sols dans les provinces des Prairies canadiennes – une menace pour la qualité du sol et pour le caractère durable de la production des écosystèmes
La salinisation est un processus typique des sols des Prairies canadiennes et des territoires adjacents des plaines boréales. On estime qu’il y a au moins 1 431 000 ha touchés par la salinisation. On distingue bien sur les deux photos ci-dessous la croûte blanche de sels à la surface du sol. On l’observe souvent en bordure des milieux humides peu profonds (marécages). La croûte est surtout constituée de sulfates de calcium et de magnésium (sels de gypse et d’Epsom) qui remontent à la surface avec l’évaporation des eaux souterraines salines que le substrat rocheux a rejetées après la période glaciaire. Un paysage où alternent douces collines et zones humides de nappe phréatique élevée favorise la formation de cette morphologie de surface.


La photo de droite montre le microrelief qui résulte du processus de salinisation dans le sol. L’effet le plus manifeste du phénomène de salinisation est sans doute le patron de croissance irrégulier que prennent les plantes de culture. La surface du sol peut sembler normale, mais la salinité est souvent plus grande en profondeur dans la zone des racines.
Après avoir été semées, les plantes germent et poussent généralement grâce à la présence d’humidité à la surface du sol. Mais à mesure que le sol sèche et que la consommation d’eau des plantes augmente, les racines pénètrent plus profondément dans le sol pour combler leur besoin en eau. Toutefois, à une certaine profondeur, elles peuvent tomber sur une concentration plus élevée de sels, laquelle peut agir comme une barrière à l’accès de l’humidité. Il en résulte un retard de croissance ou une croissance irrégulière des plantes, voire la mort faute d’humidité suffisante.
Réhabilitation des Sols Affectés par le Sel
Les méthodes d’élimination des sels de la surface du sol comprennent le raclage, le rinçage de surface et le lessivage. Étant donné la très grande solubilité des sels, la méthode du lessivage effectuée avec de l’eau de bonne qualité s’est révélé la méthode de réhabilitation des sols salins la plus efficace, par conséquent la plus utilisée. Les sols salins forment des agrégats et présentent des taux d’infiltration élevés. À l’inverse, les sols salins-sodiques et sodiques sont enclins à la dispersion et ont des taux d’infiltration faibles. Avec de telles propriétés, ces deux dernières classes de sols halomorphes se prêtent donc peu à la réhabilitation par la méthode du lessivage. C’est pourquoi on réhabilite de tels sols (salins-sodiques et sodiques) en procédant en deux étapes : d’abord par application d’amendements, tels que le gypse (CaSO4.2H2O) puis par lessivage. L’ajout de Ca2+ favorise l’agrégation du sol et améliore l’infiltration de l’eau, car le Na+ monovalent est remplacé par le Ca2+ divalent. Le Na+ ainsi retrouvé en solution avec les autres sels en excès peuvent ensuite être lessivés par irrigation avec de l’eau de bonne qualité. Cependant, pour éviter que les sels lessivés s’accumulent dans les eaux souterraines, on devra détourner cette eau saline en recourant à un drain souterrain. Il existe aussi d’autres moyens de réduire la salinité. Parce qu’ils sont en mesure d’absorber des cations et des anions dissous dans le sol, les végétaux tolérants aux sels, tels que des plantes de culture (orge, tournesol) et des arbres (peupliers, saules) peuvent faire partie de la gamme possible des méthodes de réhabilitation des sols salins. En tout état de cause, prévenir la salinisation se révèlera toujours la meilleure approche. C’est pourquoi il vaudra mieux, par exemple, toujours irriguer les terres agricoles avec de l’eau propre et recourir au drainage souterrain plutôt que d’attendre qu’elles deviennent trop concentrées en sels solubles avant d’agir.
RÉACTION D’OXYDORÉDUCTION (REDOX)
Les réactions d’oxydoréduction, ou réactions redox, font intervenir le transfert d’électrons et la conversion d’un élément d’un état de valence à un autre. Comprendre le comportement des éléments sensibles à l’oxydoréduction dans un sol, c’est aussi comprendre le comportement des sols gleysoliques et organiques et des sous-groupes de sols gleyifiés qui se trouvent dans tous les ordres de sols minéraux au Canada (SCWG, 1998). Toute l’importance de comprendre ces réactions redox se trouve justifiée par la seule production de riz, qui nécessite d’inonder temporairement des terres arables, créant les conditions propices à ces réactions. Environ 130 millions d’hectares de terres servent à la production de riz, ce qui représente environ 10 % des terres arables de la planète (IRRI, 1997).
On présente généralement les réactions redox comme des demi-réactions (c.-à-d. que l’électron et le changement d’état d’oxydation sont explicitement indiqués dans l’équation). La réaction réversible s’écrit toujours comme une réaction de réduction avec l’électron (e-) sur le côté gauche de l’équation :
oxydants + e– ↔ réducteurs
où les oxydants sont des espèces chimiques capables d’accepter des électrons et les réducteurs sont des espèces chimiques capables de donner des électrons. Par exemple :
Fe3+ + e– ↔ Fe2+
Les mesures d’oxydoréduction servent à mieux comprendre le comportement chimique des sols humides et mal drainés (p. ex. les sols gleysoliques ou organiques et les sous-groupes gleyifiés, voir la Figure 5.14) et à comprendre pourquoi un potentiel d’oxydoréduction élevé (> 300 mV) indique une bonne disponibilité d’O2 (conditions aérobiques), tandis qu’un faible potentiel redox (<300 mV) indique une disponibilité limitée d’O2 (conditions anoxiques).

Les éléments du sol les plus sensibles à l’oxydoréduction comprennent l’oxygène (O2), l’azote (NO3–, NO2–, NOx, N2O, N2), le manganése (Mn4+, Mn2+), le fer (Fe3+, Fe2+), le soufre (SO4–, S2-), et le carbone (CO2, CH4). Ce sont les éléments les plus souvent impliqués dans les réactions redox, dont la tendance décroissante à la réduction suit la séquence suivante :
Oxygéne > Azote > Manganése > Fer > Soufre > Carbone
L’état d’oxydation d’un élément va de pair avec sa tendance à la réduction, c’est-à-dire décroissante, comme l’indique la séquence ci-dessus. Plus la tendance à la réduction de l’élément est forte, plus sa capacité à accepter un électron (définie par son état d’oxydation) l’est aussi. Il s’agit d’une séquence décroissante d’acceptation d’un électron. L’élément de gauche aura préséance d’acceptation d’un électron sur les éléments de droite. Ainsi l’oxygène, en acceptant un électron, se trouve par le fait même à solliciter les éléments porteurs d’électrons, tels que certaines espèces chimiques de l’azote (NO3–, NO2–), les transformant en leur forme oxydée. Ainsi, en présence d’oxygène (c.-à-d. dans un milieu oxydant), les éléments dont la valence demeura inchangée sont le NO3–, le Mn4+, le Fe3+, le SO42-, et le CO2.
Tableau 5.5. Demi-réactions, potentiels d’électrode standard (E°), étendue habituelle du potentiel redox (Eh) dans les sols et groupes de microorganismes servant d’intermédiaire aux espèces redox dominantes dans le sol
| Demi-réaction | E° (millivolts) | Étendue du Eh (millivolts) | Microorganisme |
|---|---|---|---|
| O2(aq) + 4 H+ + 4 e- ↔ 2 H2O | 1,230 | 600 à 350 | aérobies |
| 2 NO3- + 12 H+ + 10 e- ↔ N2(g) + 6 H2O | 1,240 | 400 à 200 | de dénitrification |
| Mn(VI)O2(s) + 4 H+ + 10 e- ↔ Mn2+ + 2 H2O | 1,220 | 300 à -50 | réducteurs de manganèse |
| Fe(III)(OH)3(s) + 3 H+ + e- ↔ Fe2+ + 3 H2O | 930 | 50 à -50 | réducteurs du Fe |
| SO4 + 9 H+ + 8 e- ↔ S2- + 4 H2O | 250 | -150 à -200 | réducteurs de sulfate |
| CO2(aq) + 8 H+ + 8 e- ↔ CH4(aq) + 2 H2O | 170 | -150 à -250 | fermenteurs de méthane |
La notation avec laquelle on identifie les espèces redox dans la phase aqueuse (c.-à-d. la solution du sol) diffère de celle avec laquelle on identifie les éléments présents dans la phase solide. Par exemple, on note généralement « Fe2+ » et « Fe3+ » les espèces chimiques du fer dans la phase aqueuse, et notées « Fe(II) » et « Fe(III) » les espèces chimiques du fer contenues dans les minéraux du sol et autres solides.
En général, les microorganismes tirent leur énergie de la matière organique en la décomposant (en l’oxydant) (voir le Tableau 5.5). La matière organique est la principale source d’électrons dans le sol. Les multiples transformations/décompositions de la matière organique peuvent créer toute une gamme de composés : CO2 (gaz) résultant de la transformation totale et de composés intermédiaires (p. ex. humus) résultant de l’oxydation partielle et incomplète ; dans tous les cas, la matière organique se fait oxyder. La composition de la matière organique est d’une complexité (voir chapitre 3) telle qu’aucune formule chimique générale n’a pu à ce jour lui être attribuée (van der Park, 2006).
La Figure 5.15 présente un exemple de diagramme d’équilibres potentiel-pH, dit diagramme de Pourbaix, soit une façon de représenter l’étendue approximative des valeurs du potentiel redox (Eh) et du pH dans le sol. Les valeurs de pH varient entre 4 et 10 ; elles s’accompagnent de valeurs d’Eh, dont les limites supérieure et inférieure se trouvent définies par la stabilité redox de l’eau. Selon l’équation de Nernst, l’Eh varie en fonction du pH de -59,16 mV par unité de pH à 25 °C. La Figure 5.15 montre également l’étendue approximative des conditions oxydantes (résultant de la réaction de réduction de l’O2 dominante découlant de l’activité des organismes aérobies) ; des conditions sous-oxydantes (résultant de la réaction de réduction de l’azote, du fer et du manganèse dominante découlant de l’activité des organismes facultatifs) ; et des conditions anoxiques (résultant de la réaction de réduction du soufre et du carbone dominante découlant de l’activité des organismes anaérobies) dans le sol.
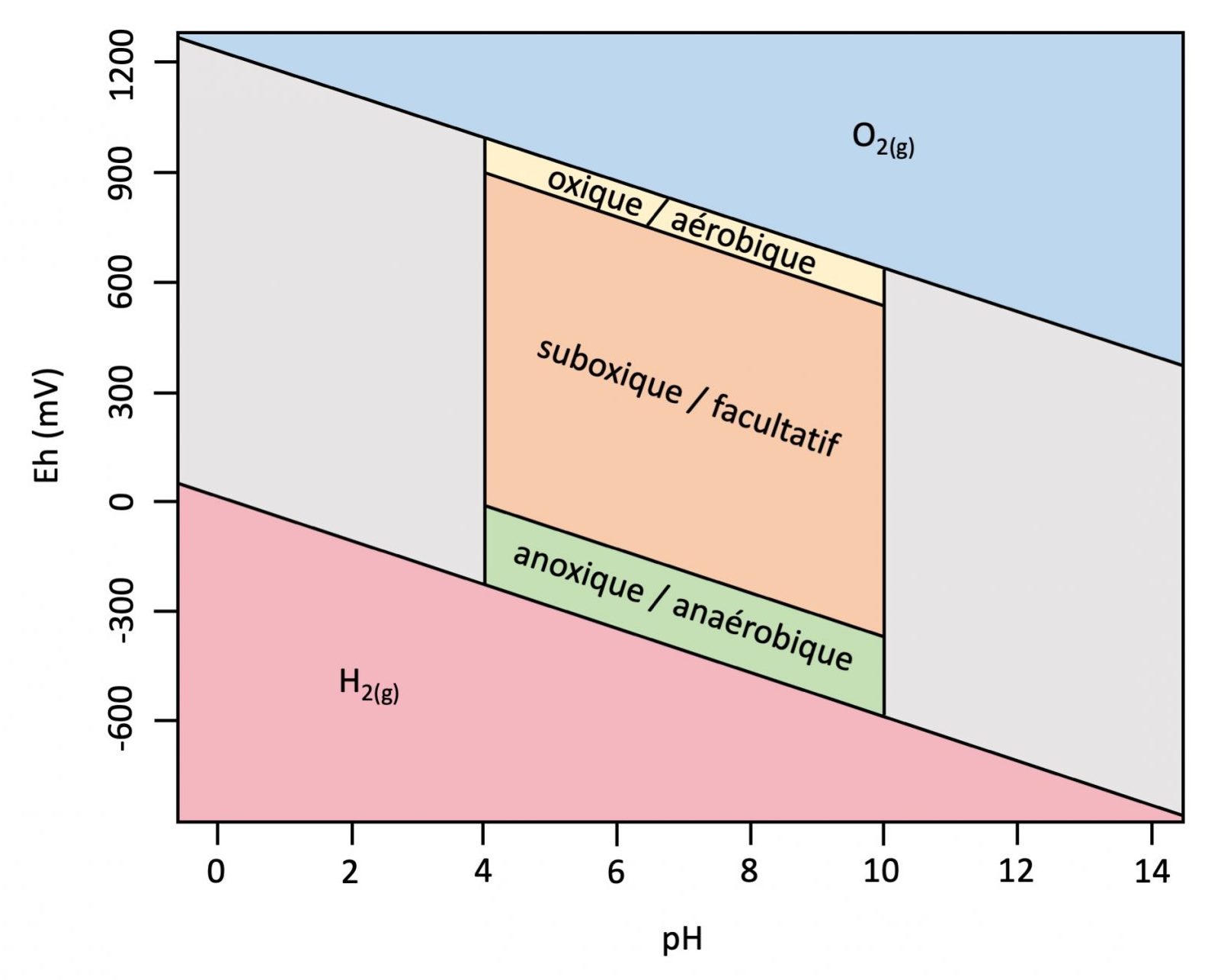
Oxygéne
Dans les sols non saturés (conditions aérobies), l’oxygène se trouve à la fois dans la phase gazeuse du sol (O2 g) et dans sa phase liquide (soit aqueuse dans la solution de sol) (O2 aq). Bien que la composition du sol soit similaire à celle de l’atmosphère (environ 20 % d’O2 en volume), dans le sol, la concentration d’oxygène dans sa phase gazeuse est généralement un peu plus faible, l’O2 se trouvant constamment consommé par les racines des plantes et les microorganismes (respiration). Dans les conditions d’un sol oxygéné (non saturé), la présence d’oxygène en quantité presque infinie est capable d’oxyder (capter les électrons) la presque totalité de la matière organique, jusqu’à l’aboutissement de CO2 (g). C’est pourquoi certaines pratiques culturales qui se trouvent à introduire de l’oxygène dans la couche arable du sol finissent par lui faire perdre sa matière organique, transformée en CO2 au terme de sa décomposition par suite des réactions d’oxydation.
Bien que la concentration d’O2 dans la phase gazeuse du sol soit généralement élevée, la concentration d’O2 dissous dans l’eau l’est moins, de l’ordre de 10-1 mg (à 15 °C), mais cette dernière peut varier suivant la température. Au fur et à mesure que l’oxygène dissous dans la solution de sol se trouve consommé, les réserves se reconstituent à partir du pool d’oxygène naturellement abondant dans la phase gazeuse du sol. Non seulement y a-t-il beaucoup plus d’oxygène dans la phase gazeuse du sol que dans sa phase liquide (solution de sol), mais l’élément y est aussi beaucoup plus mobile ; on estime la mobilité de l’oxygène à 10 000 fois plus lente dans la phase liquide que dans la phase gazeuse. Plus un sol se sature en eau, plus le transport d’oxygène ralentit, ce qui explique la diminution des valeurs d’Eh à mesure que le sol évolue vers les conditions suboxiques et même anoxiques/anaérobiques (voir fig. 5.15) (voir Figure 5.15).
Matière à réflexion !
Seasonally anoxic soils during spring snowmelt flooding

La plupart des terres agricoles sont inondées durant la période de fonte des neiges dans les Prairies canadiennes. Cette inondation saisonnière réduit le potentiel d’oxydoréduction à mesure que les conditions deviennent suboxiques ou anoxiques. Cela crée de nombreux impacts sur l’environnement. Par exemple, la concentration en phosphore dans l’eau des crues augmentera quand le Fe(III) et le Mn(IV), réduits à leur forme divalente, libéreront le phosphate qui leur est associé. Le phosphore libéré aboutira peut-être dans les lacs, ce qui favorisera la prolifération d’algues. Les conditions anoxiques entraînent aussi l’augmentation de gaz à effet de serre, tels que l’oxyde nitreux (N2O) et le méthane (CH4).
Azote
Tel qu’il vient d’être mentionné, la disponibilité en oxygène diminue rapidement dans un sol saturé en eau étant donné les quantités limitées qui s’y trouvent et la faible capacité de l’eau à le diffuser. Dans des conditions de saturation temporaire, les microorganismes réducteurs de nitrate se lancent dans le processus de dénitrification. Le processus de dénitrification ouvre la voie à la perte d’engrais azoté que l’on applique sur les cultures. Le processus fait intervenir le nitrate (NO3–) , qui se réduit en azote gazeux (N2) avec le nitrite (NO2–), générant l’oxyde nitrique soluble (NO) et l’oxyde nitreux (N2O) comme espèces intermédiaires. Le nitrite soluble est un intermédiaire aqueux à courte durée de vie ; les intermédiaires d’oxyde nitrique (NO) et d’oxyde nitreux (N2O) deux composés impliqués dans la destruction de la couche d’ozone stratosphérique (O3). L’oxyde nitreux N2O est également un puissant gaz à effet de serre, dont le potentiel de réchauffement planétaire (PRG) est de 265 à 298 fois supérieur à celui du dioxyde de carbone.
Manganèse
À mesure que le nitrate diminue, les bactéries réductrices de manganèse (Mn) prennent le relais. La teneur totale en manganèse dans le sol a beau n’être que d’environ 750 à 1000 mg kg-1 (Ure and Berrow, 1982; Bowen, 1979) il n’en constitue pas moins une espèce redox d’importance. Dans le sol, le manganèse est principalement sous forme de Mn2+ ou de Mn(VI). La forme soluble répandue dans les sols anoxiques est le Mn2+ (aq); la présence d’oxygène l’oxyde et le fait précipiter sous forme de Mn(IV) en un oxyhydroxyde non cristallin (amorphe). Les oxydes de manganèse peuvent revêtir tel un enduit noir les surfaces des particules de sol ou les couvrir de nodules noirs, certains de ces derniers pouvant mesurer jusqu’à 2 cm de diamètre. Lorsqu’ils sont présents, les solides de manganèse agissent comme des piégeurs d’anions nutritifs (p. ex. le phosphate).
Fer
Le fer est le quatrième élément en abondance dans le sol. Dans un horizon de sol, on doit les mouchetures de couleur contrastée à la valence que l’élément porte : réduite (Fe2+) et oxydée (Fe3+) laquelle valence se trouve régie par la présence ou l’absence d’eau (voir le chapitre sur la morphologie du sol). Ainsi les sols mal drainés se trouveront fortement réduits, comme l’indiqueront les valeurs d’Eh de l’ordre de -200 mV. Les horizons revêtiront la couleur grisâtre caractéristique au terme du processus de réduction du fer dans ces conditions faibles en oxygène. En effet, les conditions de réduction du sol témoignent de (1) la quasi-absence d’oxygène dissous, de nitrate et de manganèse, (2) de fer dissous qui n’est présent que sous forme d’espèces Fe2+, (3) de la plupart des composés de fer (qui confèrent à l’horizon sa couleur grise) présents sous forme d’oxydes et d’hydroxydes de Fe(II) [p. ex. Fe(OH)2]. Par suite d’un abaissement de la nappe phréatique ou d’infiltration d’eau oxygénée dans un sol réduit (peut-être par un canal racinaire, un pore ouvert ou une fracture), le fer ferreux (Fe2+) transfère ses électrons à l’oxygène, entraînant dans la foulée la formation de composés oxydés (Fe3+). Ces composés constituent les mouchetures rougeâtres facilement détectables sur le profil gris de l’horizon réduit. La présence de ces mouchetures est caractéristique des sous-groupes de sols dits à gley et des gleysols périodiquement saturés (c.-à-d. à nappe phréatique fluctuante). Les mouchetures représentent un des éléments diagnostiques des sols de drainage moyen, imparfait et mauvais.
Soufre
Bien que le soufre revête plusieurs états d’oxydation, seuls deux d’entre eux dominent dans le sol. Dans des conditions aérobies (c.-à-d. dans lesquelles l’oxygène dissous est présent), le sulfate (SO42-) constitue une forme stable du soufre, étant donné son état d’oxydation à +6, tandis que le sulfure (S2-) avec un état d’oxydation de -2 représente la forme stable dans des conditions fortement réduites (McBride, 1994). Le sulfate est la principale forme de soufre absorbée par les plantes, qui précipite principalement sous forme de gypse minéral (CaSO4•2H2O) en présence de suffisamment de calcium dissous (Ca2+) dans la solution du sol. Le sulfate peut être réduit en sulfure. Ce dernier, très faiblement soluble dans des conditions anoxiques, forme soit du sulfure d’hydrogène (H2S), reconnaissable à son odeur caractéristique d’œufs pourris, soit des sulfures métalliques insolubles, tels que FeS2 s’il y a présence de métaux solubles.
L’introduction d’oxygène dans les sols contenant des sulfures entraîne la réoxydation des sulfures métalliques :
FeS(s) + 15/4 O2 + 7/2 H2O ↔ Fe(OH)2 + 4H+ + 2 SO42-
Voilà une réaction fréquente dans les sols côtiers gagnés sur la mer, riches en sulfures, dans les sols du nord des Prairies qui dérivent de schistes sulfurés (appelés sols sulfatés acides) et dans les déchets miniers (résidus et déchets de roche) qui contiennent de la pyrite et d’autres sulfures métalliques.
Carbone
Dans l’éventualité où le sulfate arrive à sa limite dans des conditions anaérobiques, ce sont les bactéries productrices de méthane qui prennent le relais du processus de décomposition de la matière organique :
2CH2O ↔ CO2 + CH4
La formation de méthane (CH4) a surtout lieu dans les zones humides, par fermentation anaérobie de la végétation présente dans les eaux stagnantes des zones humides. Dans ces conditions anaérobiques, le processus de décomposition organique lent et incomplet entraîne l’accumulation de matière organique du sol, menant à la formation des tourbières et des sols organiques.
Mesure de la Réaction Redox
Mesurer directement le potentiel redox (réduction oxydation) dans le sol pose un vrai problème. En effet, si la mesure du potentiel redox est techniquement aussi facile à effectuer que la mesure du pH, il en est tout autre de la complexité des réactions chimiques en cause et de l’interprétation qu’il faut en tirer. Il n’y a pas de comparaison possible entre les mesures du potentiel redox — autrement dit la mesure de la « concentration d’électrons » — et les mesures du pH. La mesure du pH exprime l’activité des ions hydrogène (H+) dans la solution de sol. Or, dans la solution de sol, aucun électron impliqué dans l’activité d’oxydoréduction n’est présent. On mesure le potentiel redox en évaluant la différence de tension entre deux éléments (exprimée en volt ou en millivolt), mais cette mesure ne représente qu’une mesure du potentiel de transfert d’un électron d’un élément à un autre. Elle ne fournit qu’une indication de la capacité respective des espèces redox à recevoir un électron au moment de la mesure. Le transfert d’un électron en temps réel est beaucoup plus lent que le temps qu’il faut pour effectuer la mesure du potentiel redox. Le processus de transfert passe par une suite de réactions régies en grande partie par les microorganismes, comme il a été mentionné ci-dessus. C’est pourquoi une mesure d’oxydoréduction n’indique qu’une tendance ou qu’une probabilité qu’une réaction d’oxydoréduction se produise et dans le cas où elle se produirait, la valeur qu’on en tirerait n’aurait que celle de l’instantanéité.
En dépit des difficultés de mesurage associées au potentiel redox, il n’en demeure pas moins que les mesures d’oxydoréduction fournissent des données essentielles à la description et à l’analyse des sols qui subissent des saturations saisonnières. Les mesures se trouvent en fait des enregistrements de tension (Eh) entre une électrode de référence et une électrode de capteur insérée dans le sol. Les capteurs des électrodes les plus communes sont généralement fabriqués avec du fil de platine (électrode Pt). La tension résulte de l’échange d’électrons qui se produit au cours du processus de réduction/oxydation entre un couple oxydoréducteur, tel que le fer ferreux (Fe2+) et le fer ferrique (Fe3+) . Les sols dont les conditions de sécheresse et d’humidité fluctuent avec les saisons présentent des valeurs de potentiel d’oxydoréduction (Eh) à l’avenant (Fiedler et al., 2007).
Bien que les électrons n’existent pas à l’état libre dans un sol, le concept d’électrons libres — comme celui de protons libres (et des valeurs de pH qui lui sont associées) — se révèle utile à la description et à la compréhension de base de l’état redox. Tout comme le pH, qui exprime le degré d’activité des protons hydrogène à l’échelle logarithmique, le pe exprime aussi l’activité des électrons à l’échelle logarithmique :
(5) 
Les valeurs de pe ne se trouvent pas mesurées directement ; on doit les dériver en passant par l’équation de Nernst :
(6) 
où Eo représente le potentiel électronique standard de demi-réaction, R la constante molaire du gaz (8,31 joules/mole/K), T la température absolue (298,14 °K) ; F la constante de Faraday (96 490 coulombs/mole) et n le nombre d’électrons transférés dans la demi-réaction.
La simplicité du procédé de mesure du potentiel d’oxydoréduction (une simple électrode en platine insérée dans le sol) ne garantit pas la véracité des valeurs mesurées, car elles souffrent d’imprécisions qui découlent de beaucoup de facteurs, notamment (Sposito, 1989) :
- des valeurs de mesure, qui ne reflètent pas seulement l’activité d’oxydoréduction des demi-réactions;
- de l’incapacité de l’électrode Pt de détecter le transfert d’électrons attribuable aux espèces redox en solution, faute de leur concentration suffisante;
- du fait que beaucoup d’espèces redox ne réagissent pas facilement ;
- de la facilité de contamination des électrodes Pt ou de leur « empoisonnement » par des oxydes ou autres revêtements;
- de la différence considérable existant entre la teneur en sels de la solution de sol (et de sa capacité à conduire des électrons) et celle de la solution tampon d’étalonnage.
En raison de l’imprécision des mesures du potentiel d’oxydoréduction, on leur accorde plutôt une valeur qualitative que quantitative, qui suffit à pouvoir différencier les limites des milieux oxique, suboxique et anoxique (figure 5.15). Même si les mesures du potentiel d’oxydoréduction Eh ne peuvent constituer des mesures quantitatives des concentrations des espèces redox, elles révèlent au moins la nature des couples redox les plus actifs au moment de la mesure. Il est possible d’obtenir une mesure quantitative d’une espèce redox, à condition de la mesurer seule. Par exemple, le recours à une électrode de mesure d’un ion spécifique ou d’oxygène dissous ou le recours à un appareil de mesure multiespèces, tel que la chromatographie sur échangeur d’ions.
SOMMAIRE
-
- La chimie du sol est d’une complexité telle que pour la comprendre il faut l’étudier dans ses aspects les plus divers : composition chimique, propriétés chimiques et réactions chimiques des trois phases du sol : 1) solide (matière organique et minéraux inorganiques), 2) liquide (solution du sol) 3) gazeuse (air du sol), 4) réactions de chacune des trois phases (composante non biotique du sol) avec les plantes et les organismes qui y vivent (composante biotique du sol).
Le bon fonctionnement des propriétés du sol assure la santé des humains et de l’environnement de toute la planète. Les propriétés du sol soumises à l’influence des apports d’engrais et des contaminants exercent un rôle moteur sur une vaste gamme d’activités humaines (production des cultures, gestion de la sécurité alimentaire) et de ses effets sur l’environnement (modification de la biodiversité, pollution de l’air et de l’eau, émissions de gaz à effet de serre, etc.).
L’acquisition des connaissances sur la chimie du sol se révèle fondamentale à la compréhension des effets de ces apports d’engrais et des contaminants sur les sols de sorte que l’on puisse non seulement en prévoir les impacts sur l’environnement, mais aussi trouver des façons de les réduire. - Les propriétés chimiques du sol subissent l’influence du type et de la quantité de minéraux présents (matière inorganique) dans le sol et de la teneur en matière organique (humus). Les colloïdes du sol renvoient à la fraction des particules du sol <0,002 mm. Les particules peuvent être autant d’origine inorganique qu’organique. Les colloïdes inorganiques comptent les aluminosilicates du type cristallin (phyllosilicates) et du type amorphe et les minéraux oxyhydroxydes.
- Les charges de surface sur les colloïdes de matière inorganique (minéraux) et organique (humus) sont en cause dans le processus d’adsorption et d’échange d’ions dans les sols. La plupart des charges de surface des particules sont dites permanentes (c.-à-d. indépendantes des variations de pH du sol) et négatives ; d’autres particules ont des charges de surface qui varient avec le pH et peuvent être négatives ou positives.
- Les colloïdes du sol chargés négativement attirent les cations, les adsorbent, puis peuvent les libérer dans la solution de sol au cours d’un processus d’échange d’ions. La capacité d’échange cationique (CEC) se définit comme étant la quantité totale de cations échangeables qu’un sol peut adsorber. La capacité du sol à « retenir » les cations sert les plantes qui s’en nourrissent et sert aussi à retenir les cations polluants qui seraient autrement lessivés.
- Le pH du sol est une mesure de l’activité des ions hydrogène dans le sol, qui exprime son caractère acide pH < 7, neutre (pH = 7) ou alcalin (pH > 7). Le pH régit nombre de processus chimiques qui ont lieu dans le sol, y compris celui qui régit la disponibilité des éléments nutritifs nécessaires aux plantes. Le principal facteur d’influence du pH d’un sol est la nature du matériau géologique d’origine dont il provient. Le chaulage est une pratique agricole courante qui consiste à apporter des amendements calciques en vue de corriger l’acidité du sol (faire remonter le pH).
- La salinité d’un sol définit la quantité de sels solubles dans un sol, que l’on détermine en mesurant la conductivité électrique d’une solution de sol qui a été extraite d’une pâte de sol saturé. On distingue trois classes de sols halomorphes (sols touchés par la salinisation) en fonction de la concentration relative de Na+ présente dans la solution de sol ou sur le complexe d’échange : les salins, les salins-sodiques et les sodiques. Irriguer avec de l’eau saline, particulièrement dans les climats arides, constitue une cause anthropique majeure de salinisation. Un excès de salinité dans le sol peut faire diminuer la disponibilité de l’eau pour les plantes, par conséquent affecter leur croissance.
- Les réactions d’oxydoréduction (redox) dans le sol impliquent la conversion d’un élément d’un état de valence à un autre. Les éléments du sol les plus sensibles à l’oxydoréduction comprennent l’oxygène (O2), l’azote (NO3–, NO2–, NOx, N2O, N2), manganèse (Mn4+, Mn2+), fer (Fe3+, Fe2+), soufre (SO42-, S2-), et carbone (CO2, CH4). Les réactions d’oxydoréduction sont en cause dans la dynamique de certains éléments nutritifs des plantes (p. ex. l’azote, le soufre, le fer et le manganèse) et dans la production de gaz qui s’échappent des sols, y compris les gaz à effet de serre, tels que l’oxyde nitreux et le méthane.
- La chimie du sol est d’une complexité telle que pour la comprendre il faut l’étudier dans ses aspects les plus divers : composition chimique, propriétés chimiques et réactions chimiques des trois phases du sol : 1) solide (matière organique et minéraux inorganiques), 2) liquide (solution du sol) 3) gazeuse (air du sol), 4) réactions de chacune des trois phases (composante non biotique du sol) avec les plantes et les organismes qui y vivent (composante biotique du sol).
EXERCICES PRATIQUES ET EXAMPLES D’ETUDE DE SOL
- Expliquez pourquoi la montmorillonite a une plus grande capacité de gonflement que la kaolinite.
- Donnez la définition de la substitution isomorphe. Expliquez brièvement comment la substitution isomorphe déclenche l’apparition de charges négatives de surface sur les surfaces des minéraux argileux.
- Expliquez pourquoi les vertisols au Canada sont généralement très collants lorsqu’ils sont humides et forment de larges fissures lorsqu’ils sont secs.
- Donnez la définition de la capacité d’échange cationique. Quelle est la part de rôle que joue la capacité d’échange cationique du sol dans la protection et l’amélioration de la biodiversité et de la santé des écosystèmes ?
- En vous aidant de la figure 5.2 sur la structure et les propriétés des phyllosilicates, répondez aux questions Vrai / Faux.
- La présence de liaisons hydrogène intercouches fait de la kaolinite un minéral argileux qui gonfle peu. Vrai ou Faux
- Les minéraux argileux de type 2:1 sont composés d’un feuillet tétraédrique pris en sandwich entre deux feuillets octaédriques. Vrai ou Faux
- Il n’y a pas de liaisons hydrogène entre deux couches dans les minéraux argileux de type 2:1, car les groupes hydroxyles ne sont pas exposés. Vrai ou Faux
- La montmorillonite et l’illite (mica hydraté) sont toutes deux des phyllosilicates de type 2:1. La montmorillonite gonfle librement, tandis que l’illite a une capacité de gonflement limitée.
Vrai ou Faux - L’illite (mica hydraté) a une charge de couche unitaire élevée en raison de la substitution isomorphe, mais sa capacité d’échange cationique demeure relativement faible. Vrai ou Faux
- Un sol possédant une capacité d’échange cationique de 42 cmol(+) kg-1 est doté des cations suivants sur le complexe d’échange : Na+ = 7 cmol(+) kg-1 de sol, Ca2+= 12 cmol(+) kg-1 de sol, K+= 6 cmol(+) kg-1 de sol, et Mg2+= 8 cmol (+) kg-1 de sol). Calculez sa saturation en base.
- Expliquez en quoi le fait de modifier le pH d’un sol influence ses capacités d’échange de cations et d’anions.
- Quelle est la différence entre l’acidité active et l’acidité d’échange ?
- « Le principal facteur d’influence du pH d’un sol est la nature du matériau d’origine dont il provient ». Étayez votre réponse en donnant des exemples de matériau d’origine au Canada.
- Expliquez la façon dont les colloïdes du sol tamponnent les changements de pH du sol causés par des intrants acides ou basiques, tels que les engrais contenant de l’azote et les amendements contenant de la chaux.
- Quelle est l’influence du pH sur la disponibilité des éléments nutritifs pour les plantes ?
- Donnez la définition des sols salins, salins-sodiques et sodiques. Relevez leurs principales caractéristiques et leurs effets sur la santé des écosystèmes.
- En quoi la salinité d’un sol dans une région au climat aride se trouve-t-elle influencée par l’irrigation ?
- Un sol d’une capacité d’échange cationique de 20 cmol(+) kg-1 a une teneur en Na échangeable de 8 cmol(+) kg-1 :
- Calculez le pourcentage de sodium échangeable (PSE) du sol.
- Si la conductivité électrique de l’extrait saturé de ce sol est de 5,6 dS m-1 (deciSiemens par mètre), à quelle classe appartient-il ? (indice: voir Figure 5.11)
- Peut-on réhabiliter un tel sol ? Si oui, comment ?
- Énumérez les principaux éléments sensibles à l’oxydoréduction dans le sol. Donnez une forme oxydée et une forme réduite de chacun d’eux.
- L’état redox du sol influence la disponibilité des éléments nutritifs et les émissions de gaz à effet de serre (oxyde nitreux et méthane). Expliquez.
EXAMPLES D’ETUDE DE SOL
Décrivez les mécanismes qui régissent l’apparition des charges négatives sur les surfaces des colloïdes du sol
Deux grands mécanismes régissent les charges négatives sur les surfaces des colloïdes du sol.
Le premier mécanisme en est un de substitution, dite isomorphe. La substitution isomorphe porte sur les cations constitutifs du centre des feuillets tétraédriques ou octaédriques d’un minéral phyllosilicaté, qui se font remplacer par des cations de taille similaire. Le qualificatif isomorphe indique que la substitution ne modifie pas la structure cristalline. Dans le cas où le cation de remplacement est de valence inférieure à celui qui vient d’être remplacé, la charge de surface résultante est négative. Par exemple, le remplacement de Si4+ par l’Al3+ dans le feuillet tétraédrique (comme dans l’illite ou le mica hydraté) conférera à la surface du colloïde une charge négative. De même, dans le feuillet octaédrique, si Al3+ se fait remplacer par Mg2+, la surface du colloïde devient chargée négativement. Le mécanisme de substitution isomorphe produit des charges de surface négatives permanentes, qui demeurent indépendantes des variations du pH dans le sol. De même, dans le feuillet octaédrique, si Al3+ se fait remplacer par Mg2+ la surface du colloïde devient chargée négativement.
Le deuxième mécanisme d’apparition de charges négatives sur les colloïdes, autant les organiques que les inorganiques, est tributaire du pH. Certaines valeurs de pH déclenchent le mécanisme dit de protonation, qui se trouve à faire perdre aux colloïdes des protons, rendant leur charge de surface négative. Ainsi en est-il des argiles oxyhydroxydes, des argiles phyllosilicates (bords) et des silicates amorphes, dont les groupes hydroxyles peuvent perdre un proton à pH élevé. De même à pH élevé, les surfaces des groupes fonctionnels des colloïdes organiques (humus), tels que les groupes carboxylés et phénoliques peuvent être porteuses de charges négatives par déprotonation.
La montmorillonite minérale argileuse est dotée d’une CEC plus élevée que celle de la kaolinite. Expliquez (indice : voir Figure 5.2)
Les différences de CEC découlent des différences de structure entre les deux argiles phyllosilicates. La montmorillonite est un phyllosilicate de structure du type 2:1, qui est formée de 2 feuillets tétraédriques de silicium entre lesquels se trouve un feuillet octaédrique d’aluminium. Par substitution isomorphe, par exemple, par le remplacement de l’aluminium (Al3+) par du magnésium (Mg2+) dans un feuillet octaédrique, la charge de surface du colloïde de montmorillonite devient porteuse d’une charge négative permanente. De plus, les argiles montmorillonites ont la propriété de gonfler lorsque de l’eau pénètre dans l’espace (dit espace intercouche) qui sépare les deux feuillets tétraédriques de deux couches unitaires 2:1 juxtaposées. Or, cet ajout de surface spécifique disponible favorise l’adsorption des cations, car dans de telles conditions autant les surfaces externes et internes de la montmorillonite peuvent adsorber des cations. La capacité de la montmorillonite de créer des charges négatives nettes par substitution isomorphe et d’augmenter sa surface spécifique dans des conditions d’humidité fait d’elle une argile à CEC élevée.
La kaolinite est un minéral d’argile phyllosilicate du type 1:1, qui se compose de feuillets tétraédriques simples de silicium qui alternent avec des feuillets octaédriques d’aluminium simples. De par la structure de type 1:1 de la kaolinite, le phénomène d’adsorption de cations par substitution isomorphe ne peut avoir lieu, limitant du coup sa possibilité de créer des charges négatives permantes. De plus, l’absence d’espace intercouche de la structure de type 1:1 de la kaolinite ne lui permet pas de gonfler (comme c’est le cas de la montmorillonite), limitant par conséquent la surface spécifique sur laquelle des cations pourraient être adsorbés. On en conclut que la structure de type 1:1 explique pourquoi la kaolinite a une CEC plus faible que celle de la montmorillonite, en raison du fait : 1) qu’elle ne se prête pas au phénomène de substitution isomorphe et 2) que son incapacité de gonfler l’empêche d’augmenter la surface spécifique sur laquelle se produisent les échanges de cations.
Indiquez les caractéristiques des phyllosilicates dans le tableau ci-dessous
| Minéral | Type | Substitution isomorphe | CEC | Surface spécifique | Capacité de gonflement |
|---|---|---|---|---|---|
| (1:1, 2:1, 2:1:1) | (faible/élevée) | (faible/élevée) | (faible/élevée) | (faible/élevée) | |
| Kaolinite | 1:1 | faible | faible | faible | faible |
| Montmorillonite | 2:1 | élevée | élevée | élevée | élevée |
| Illite (mica hydraté) | 2:1 | élevée | faible | faible | faible |
| Chlorite | 2:1:1 | élevée | faible | faible | faible |
Calculez le CEC du sol
Un sol contient 3 % de matière organique, 20 % de montmorillonite et 17 % de kaolinite ; les 60 % restants étant composés de grains de minéraux primaires de sable et de limon. Calculer la capacité d’échange cationique du sol en cmol (+) kg-1 (Remarque : supposons que la capacité d’échange cationique de la matière organique = 200 cmol(+) kg-1,, la montmorillonite = 100 cmol(+) kg-1 et la kaolinite = 10 cmolc(+) kg-1).
Le CEC du sol = [0.03 x 200 cmolc(+) kg-1 ] + [0.20 x 100 cmolc(+) kg-1] + [0.17 x 10 cmol (+) kg-1] = 27,7 cmolc(+) kg-1
Nommez les quatre principaux facteurs qui contribuent à l’acidité du sol. Expliquez brièvement leur contribution à l’acidité
Les quatre facteurs qui contribuent à l’acidité du sol sont le matériau parental d’origine acide, l’apport des précipitations, l’application d’engrais à l’ammonium et la décomposition de la matière organique. Les sols dérivés de matériaux d’origine granitique, communément appelés matériaux d’origine acides, ont des réactions qui génèrent de l’acidité. L’acidité naturelle des précipitations attribuable à la présence de dioxyde de carbone dissous participe à l’acidification du sol. Dans les zones de précipitations intenses, les cations basiques dans les sols se font lessiver, ce qui a pour effet d’augmenter leur acidité. L’application d’engrais à l’azote ammoniacal peut également contribuer à l’acidification, car la nitrification de l’ammonium libère des protons. La décomposition de la matière organique libère des acides organiques apportant sa part d’acidité du sol.
Nommez deux causes naturelles et deux causes anthropiques de salinité du sol et décrivez brièvement le rôle qu’elles jouent sur son augmentation
Deux causes naturelles de la salinité d’un sol : des conditions climatiques arides, un mauvais drainage. Dans les conditions arides, les sels solubles s’accumulent faute de précipitations suffisantes pour les lessiver adéquatement. Ces mêmes conditions arides font perdre de l’eau aux sols par évaporation. Il se produit alors un mouvement ascendant de l’eau des couches profondes du sol vers la surface, entraînant avec lui les sels solubles qui resteront à la surface.
Dans des conditions de mauvais drainage (en raison de la nappe phréatique élevée ou de la faible perméabilité du sous-sol), les sels ne s’éliminent pas par drainage et lessivage, ce qui les fait s’accumuler à la surface une fois l’eau évaporée.
Deux causes anthropiques de salinité des sols : l’irrigation avec de l’eau saline (en régions sèches), surutilisation d’engrais. L’eau d’irrigation dissout plus de sels par écoulement naturel vers le bas du profil de sol. Dans cette situation, les conditions d’évaporation élevée attribuable au climat sec apporteront des sels dissous à la surface du sol sous l’effet de la remontée d’eau. Les engrais (qui sont en fait des sels contenant divers éléments nutritifs) peuvent augmenter la teneur en sels des sols s’ils sont utilisés de manière inappropriée.
Soit les données suivantes portant sur 4 sols minéraux qui ont tous une texture de loam argileux :
| Propriété du sol | Sol n° 1 | Sol n° 2 | Sol n° 3 | Sol n° 4 |
|---|---|---|---|---|
| pH | 8,8 | 8,1 | 7,5 | 7,6 |
| Conductivité électrique (dS/m) | 2,7 | 12,2 | 1 | 4,3 |
| Saturation en bases (%) | 90 | 90 | 60 | 86 |
| Capacité d’échange cationique (cmolc/kg) | 12 | 11 | 19 | 9 |
| Pourcentage de sodium échangeable ou PSE | 17,3 | 16,8 | 2,1 | 2,5 |
| Classe de salinité | sodique | salin-sodique | non salin | salin |
A) Déterminez la classe de salinité (salin, sodique, salin-sodique, non salin) de chacun de ces 4 sols. [indice : voir Figure 5.12]
B) Lequel des quatre sols serait celui doté de la propriété de dispersion la plus élevée ? Expliquez brièvement votre réponse en vous basant sur les informations fournies.
Le sol n° 1 est fort probablement celui qui a la plus forte capacité de dispersion, étant donné son PSE de 17,3 % et sa faible concentration en sels (CE de 2,7 dS m-1).
C) Quels sont les impacts de cette propriété de dispersion sur la croissance des plantes ?
La dispersion des particules favorise la formation de micropores peu propices au mouvement de l’air et de l’eau, des conditions défavorables à la croissance des racines des plantes, par conséquent de la croissance des plantes elles-mêmes.
Déterminez les formes oxydées et réduites communes de ces éléments dans les sols : manganèse, fer, soufre, and azote
- manganèse : Mn4+, Mn2+ (reduced)
- fer : Fe3+, Fe2+
- soufre : SO42-, S2-
- azote : NO3–, NO2–, NOx, N2O, N2
RÉFÉRENCES
Acton D.F., and Gregorich L.J. 1995. The health of our soils: toward sustainable agriculture in Canada. Agriculture Agri-Food Canada, CDR Unit, Ottawa.
Bowen H.J.M. 1979. Environmental chemistry of the elements. Academic Press, London, New York.
Brady N.C. and Weil, R.R. 2010. The Nature and Properties of Soils 13th Ed. Pearson Education.
Canada Department of Agriculture; Research Branch 1976. Glossary of Terms in Soil Science, Publication 1459.
Eilers W., MacKay R., Graham L. and Lefebvre A. (eds.). 2010. Environmental Sustainability of Canadian Agriculture: Agri-Environmental Indicator Report Series — Report #3. Agriculture and Agri-Food Canada, Ottawa, Ontario.
FAO 2020. Food and Agriculture Organization of the United Nations. FAO Soil Portal. http://www.fao.org/soils-portal/en/
Fiedler S., Vepraskas M.J., and Richardson J.L. 2007. Soil Redox Potential: Importance, Field Measurements and Observations. Advances in Agronomy 94:1-54.
Havlin J.L., Tisdale S.L, Nelson W.L., and Beaton J.D. 2013. Soil Fertility and Fertilizers: An Introduction to Nutrient Management, 8th ed. Upper Saddle River, NJ: Prentice Hall.
Hendershot W.H., Lalande H., and Duquette M. 2008. Soil Reaction and Exchangeable Acidity. Pages 173-178 IN Carter, M.R. and Gregorich, E.G. (eds.) Soil Sampling and Methods of Analysis. 2nd Edition. Canadian Society of Soil Science. CRC Press, Baton Rouge.
Hendershot W.H., Lalande H., and Duquette M. 1993. Soil Reaction and Exchangeable Acidity. Pp. 167-176 in Carter M.R. (ed.) Soil Sampling and Methods of Analysis. 1st Edition. Canadian Society of Soil Science. CRC Press, Baton Rouge.
International Rice Research Institute (IRRI). 1997. Rice Almanac, 2nd Ed. International Rice Research Institute, Manila.
McKenzie R.H. and Woods S.A. 2010. Management of sodic soils in Alberta, Agdex 518-20, Alberta Agriculture. https://www1.agric.gov.ab.ca/$department/deptdocs.nsf/all/agdex13200/$file/518-20.pdf?Open Element
McBride M.B. 1994. Environmental Chemistry of Soils. Oxford University Press. New York. 406pp.
Miller J.J. and Brierley J.A. 2011. Solonetzic soils of Canada: Genesis, distribution and classification. Can. J. Soil Sci. 91(5):889-902.
Munroe J. (ed.) 2018. Soil Fertility Handbook. Publication 611 3rd edition, Ontario Ministry of Agriculture, Food and Rural Affairs. 236pp.
Newman A.C. and Hayes M.H. 1990. Some properties of clays and of other soil colloids and their influences on soils. In: De Boodt M.F, M.H. Hayes, A. Herbillon (Eds). Soil colloids and their associations in aggregates. Springer Science & Business Media; Boston. MA, USA. pp. 39-55.
Qadir M., Quillérou E., Nangia V., Murtaza G., Singh M., Thomas R.J., Drechsel P., and Noble A.D. 2014. Economics of salt‐induced land degradation and restoration. In: Natural Resources Forum 38 (4):282-295.
Shreeja D. (n.d.) Phosphate Fixation in Soil: 3. Reactions of Anion Fixation. Available at https://www.soilmanagement india.com/nutrient-elements-in-soil/phosphate-fixation-in-soil-3-reactions-anion-fixation/2252
Sparks D.L. 2003. Environmental Soil Chemistry. Academic Press, London, UK.
Sparks D.L. 2019. Fundamentals of Soil Chemistry. Encyclopedia of Water: Science, Technology, and Society, pp 1-11.
Sposito G. 1989. The Chemistry of Soils. Oxford University Press. New York. 277pp.
Soil Classification Working Group (SCWG), 1998. The Canadian System of Soil Classification. 3rd Edition, Research Branch Agriculture and Agri-Food Canada, Publication 1646.
Soil Survey Staff. 2014. Kellogg Soil Survey Laboratory Methods Manual. Soil Survey Investigations Report No. 42, Version 5.0. R. Burt and Soil Survey Staff (ed.). U.S. Department of Agriculture, Natural Resources Conservation Service. 1001pp.
Ure A.M., and Berrow M.L. 1982. The Elemental Constituents in Soils. Pages 94-204 IN Bowen, H.J.M. (ed.). Environmental Chemistry. Volume 2. Royal Soc. Chem. London.
Van der Perk M. 2006. Soil and Water Contamination from Molecular to Catchment Scale. Taylor and Francis, London 389pp.
Zheng T. 2014. Numerical Analysis of Modeling Concepts for Salt Precipitation and Porosity-Permeability Evolution during Brine Evaporation. Doctoral dissertation, Universität Stuttgart, Germany.
Les Auteurs
Darshani Kumaragamage, Professeure, Département des sciences et des études environnementales, Université de Winnipeg, MB

J’ai obtenu un baccalauréat ès sciences et une maîtrise en agriculture de l’Université de Peradeniya, au Sri Lanka, et un doctorat en sciences du sol de l’Université du Manitoba, au Canada, spécialisé en chimie physique des sols. J’ai plus de 30 ans d’expérience dans l’enseignement au niveau universitaire et j’ai enseigné une variété de cours en sciences du sol, notamment l’introduction à la science des sols, la chimie des sols et la fertilité des sols, ainsi que des cours en sciences de l’environnement. Mes intérêts de recherche comprennent l’évaluation, la surveillance et l’atténuation des impacts environnementaux des engrais et du fumier animal, avec un accent particulier sur les transformations et la mobilité du phosphore dans les sols sous différents scénarios climatiques et de gestion. J’ai écrit/coécrit 65 publications dans des revues à comité de lecture.
Mon souvenir préféré de travail avec le sol est celui où j’ai mené une recherche sur le terrain au Manitoba sur un sol argileux très collant, chernozémique, un jour de pluie. J’ai dû prélever des échantillons de sol sur de nombreuses parcelles, et alors que j’entrais dans le champ, environ un kilo de terre s’est collé à mes bottes. Avec le pas suivant, ça a presque doublé, et à chaque pas que je faisais, mes bottes devenaient de plus en plus lourdes jusqu’à ce que je doive littéralement traîner les pieds pour passer d’une parcelle à l’autre.
C. James (Jim) Warren, Ph.D. P.Géo., spécialiste en ressources foncières, ministère de l’Agriculture, de l’Alimentation et des Affaires rurales de l’Ontario, Guelph, ON

Je suis géo. et un pédologue environnementaliste avec une expérience diversifiée dans la recherche, le conseil et l’enseignement. Je suis diplômé de l’Université de Guelph (B.Sc. (Agr), Sciences du sol) et de l’Université de l’Alberta (M.Sc. Science des sols; Ph.D. Chimie des sols). J’ai mené des recherches en agronomie, géochimie et minéralogie à l’Université de Guelph et à l’Université de Waterloo avant de devenir consultant privé en drainage minier acide. Je suis actuellement employé en tant que spécialiste des ressources foncières qui effectue des études de sol et des travaux de pédologie avec le ministère de l’Agriculture, de l’Alimentation et des Affaires rurales de l’Ontario, et je suis également professeur auxiliaire à l’Université de Guelph. J’ai donné des cours en science du sol, minéralogie du sol, chimie du sol, géochimie et pédologie en tant que chargé de cours dans quatre universités (Alberta, Guelph, Waterloo et Toronto). J’ai écrit ou co-écrit plus de 100 publications dans des revues à comité de lecture ; chapitres de livres, actes de conférences et rapports.
Comment me suis-je intéressé aux sols ? Eh bien, j’ai grandi dans une ferme où, adolescent, je passais beaucoup de temps sur un tracteur à faire des travaux de labour et j’avais beaucoup de temps pour réfléchir. Une des questions que je me suis posée pendant de longues journées sur le terrain était : « Qu’est-ce qui fait pousser les plantes ? Avec cela, j’ai passé les décennies suivantes à essayer de comprendre les sols pour répondre à cette question. La quête continue . . .
Graeme Spiers, directeur de la surveillance environnementale et professeur, École de l’environnement, Département des sciences de la Terre / Département de biologie, Université Laurentienne, Sudbury, ON

J’enseigne à l’École de l’environnement, au Département des sciences de la Terre et au Département de biologie de l’Université Laurentienne. J’ai obtenu un B.Sc. en Sciences de la Terre et Botanique à l’Université de Waikato en Nouvelle-Zélande, et M.Sc. et Ph.D. diplômes de l’Université de l’Alberta, spécialisés en pédologie, minéralogie de l’argile et chimie. Avec une expérience de laboratoire commercial et de consultation à travers le Canada, j’ai été associé à des programmes de recherche aux universités de l’Alberta, de Guelph, de Waterloo, des Laurentides et du Nouveau-Brunswick. Mes recherches ont donné lieu à de nombreuses publications et présentations techniques et scientifiques en chimie analytique, chimie aquatique, géologie environnementale, écologie, biologie végétale et science du sol. Avant d’aller à l’université à temps plein, j’ai été éleveur laitier néo-zélandais pendant 10 ans, obtenant mon diplôme de premier cycle comme passe-temps à long terme entre les traites. À Sudbury, je participe à des initiatives de recherche examinant les effets des émissions de métaux anthropiques historiques et actuelles sur les sols, les rivières et les lacs, ainsi que la végétation dans l’empreinte de la fonderie de Sudbury. Je suis activement impliqué dans une variété de programmes de recherche à travers le Canada et collabore activement avec des chercheurs en Russie (Université d’État de Moscou et Académie des sciences de Russie), en Australie, en Nouvelle-Zélande, en Chine, au Pérou et en Afrique du Sud.
