Chapter 4. Genetics and Evolution
4.3 Epigenetics in Psychology
Ian Weaver
Early life experiences exert a profound and long-lasting influence on physical and mental health throughout life. The efforts to identify the primary causes of this have significantly benefited from studies of the epigenome—a dynamic layer of information associated with DNA that differs between individuals and can be altered through various experiences and environments. The epigenome has been heralded as a key “missing piece” of the etiological puzzle for understanding how development of psychological disorders may be influenced by the surrounding environment, in concordance with the genome. Understanding the mechanisms involved in the initiation, maintenance, and heritability of epigenetic states is thus an important aspect of research in current biology, particularly in the study of learning and memory, emotion, and social behavior in humans. Moreover, epigenetics in psychology provides a framework for understanding how the expression of genes is influenced by experiences and the environment to produce individual differences in behavior, cognition, personality, and mental health. In this module, we survey recent developments revealing epigenetic aspects of mental health and review some of the challenges of epigenetic approaches in psychology to help explain how nurture shapes nature.
Learning Objectives
- Explain what the term epigenetics means and the molecular machinery involved.
- Name and discuss important neural and developmental pathways that are regulated by epigenetic factors, and provide examples of epigenetic effects on personality traits and cognitive behavior.
- Understand how misregulation of epigenetic mechanisms can lead to disease states, and be able to discuss examples.
- Recognize how epigenetic machinery can be targets for therapeutic agents, and discuss examples.
Introduction

Early childhood is not only a period of physical growth; it is also a time of mental development related to changes in the anatomy, physiology, and chemistry of the nervous system that influence mental health throughout life. Cognitive abilities associated with learning and memory, reasoning, problem solving, and developing relationships continue to emerge during childhood. Brain development is more rapid during this critical or sensitive period than at any other, with more than 700 neural connections created each second. Herein, complex gene–environment interactions (or genotype–environment interactions, G×E) serve to increase the number of possible contacts between neurons, as they hone their adult synaptic properties and excitability. Many weak connections form to different neuronal targets; subsequently, they undergo remodeling in which most connections vanish and a few stable connections remain. These structural changes (or plasticity) may be crucial for the development of mature neural networks that support emotional, cognitive, and social behavior. The generation of different morphology, physiology, and behavioral outcomes from a single genome in response to changes in the environment forms the basis for “phenotypic plasticity,” which is fundamental to the way organisms cope with environmental variation, navigate the present world, and solve future problems.
The challenge for psychology has been to integrate findings from genetics and environmental (social, biological, chemical) factors, including the quality of infant–mother attachments, into the study of personality and our understanding of the emergence of mental illness. These studies have demonstrated that common DNA sequence variation and rare mutations account for only a small fraction (1%–2%) of the total risk for inheritance of personality traits and mental disorders (Dick, Riley, & Kendler, 2010; Gershon, Alliey-Rodriguez, & Liu, 2011). Additionally, studies that have attempted to examine the mechanisms and conditions under which DNA sequence variation influences brain development and function have been confounded by complex cause-and-effect relationships (Petronis, 2010). The large unaccounted heritability of personality traits and mental health suggests that additional molecular and cellular mechanisms are involved.
Epigenetics has the potential to provide answers to these important questions and refers to the transmission of phenotype in terms of gene expression in the absence of changes in DNA sequence—hence the name epi- (Greek: επί- over, above) genetics (Waddington, 1942; Wolffe & Matzke, 1999). The advent of high-throughput techniques such as sequencing-based approaches to study the distributions of regulators of gene expression throughout the genome led to the collective description of the “epigenome.” In contrast to the genome sequence, which is static and the same in almost all cells, the epigenome is highly dynamic, differing among cell types, tissues, and brain regions (Gregg et al., 2010). Recent studies have provided insights into epigenetic regulation of developmental pathways in response to a range of external environmental factors (Dolinoy, Weidman, & Jirtle, 2007). These environmental factors during early childhood and adolescence can cause changes in expression of genes conferring risk of mental health and chronic physical conditions. Thus, the examination of genetic–epigenetic–environment interactions from a developmental perspective may determine the nature of gene misregulation in psychological disorders.
This module will provide an overview of the main components of the epigenome and review themes in recent epigenetic research that have relevance for psychology, to form the biological basis for the interplay between environmental signals and the genome in the regulation of individual differences in physiology, emotion, cognition, and behavior.
Molecular control of gene expression: the dynamic epigenome

Almost all the cells in our body are genetically identical, yet our body generates many different cell types, organized into different tissues and organs, and expresses different proteins. Within each type of mammalian cell, about 2 meters of genomic DNA is divided into nuclear chromosomes. Yet the nucleus of a human cell, which contains the chromosomes, is only about 2 μm in diameter. To achieve this 1,000,000-fold compaction, DNA is wrapped around a group of 8 proteins called histones. This combination of DNA and histone proteins forms a special structure called a “nucleosome,” the basic unit of chromatin, which represents a structural solution for maintaining and accessing the tightly compacted genome. These factors alter the likelihood that a gene will be expressed or silenced. Cellular functions such as gene expression, DNA replication, and the generation of specific cell types are therefore influenced by distinct patterns of chromatin structure, involving covalent modification of both histones (Kadonaga, 1998) and DNA (Razin, 1998).
Importantly, epigenetic variation also emerges across the lifespan. For example, although identical twins share a common genotype and are genetically identical and epigenetically similar when they are young, as they age they become more dissimilar in their epigenetic patterns and often display behavioral, personality, or even physical differences, and have different risk levels for serious illness. Thus, understanding the structure of the nucleosome is key to understanding the precise and stable control of gene expression and regulation, providing a molecular interface between genes and environmentally induced changes in cellular activity.
The primary epigenetic mark: DNA modification
DNA methylation is the best-understood epigenetic modification influencing gene expression. DNA is composed of four types of naturally occurring nitrogenous bases: adenine (A), thymine (T), guanine (G), and cytosine (C). In mammalian genomes, DNA methylation occurs primarily at cytosine residues in the context of cytosines that are followed by guanines (CpG dinucleotides), to form 5-methylcytosine in a cell-specific pattern (Goll & Bestor, 2005; Law & Jacobsen, 2010; Suzuki & Bird, 2008). The enzymes that perform DNA methylation are called DNA methyltransferases (DNMTs), which catalyze the transfer of a methyl group to the cytosine (Adams, McKay, Craig, & Burdon, 1979). These enzymes are all expressed in the central nervous system and are dynamically regulated during development (Feng, Chang, Li, & Fan, 2005; Goto et al., 1994). The effect of DNA methylation on gene function varies depending on the period of development during which the methylation occurs and location of the methylated cytosine. Methylation of DNA in gene regulatory regions (promoter and enhancer regions) usually results in gene silencing and reduced gene expression (Ooi, O’Donnell, & Bestor, 2009; Suzuki & Bird, 2008; Sutter and Doerfler, 1980; Vardimon et al., 1982). This is a powerful regulatory mechanism that ensures that genes are expressed only when needed. Thus DNA methylation may broadly impact human brain development, and age-related misregulation of DNA methylation is associated with the molecular pathogenesis of neurodevelopmental disorders.
Histone modification and the histone code
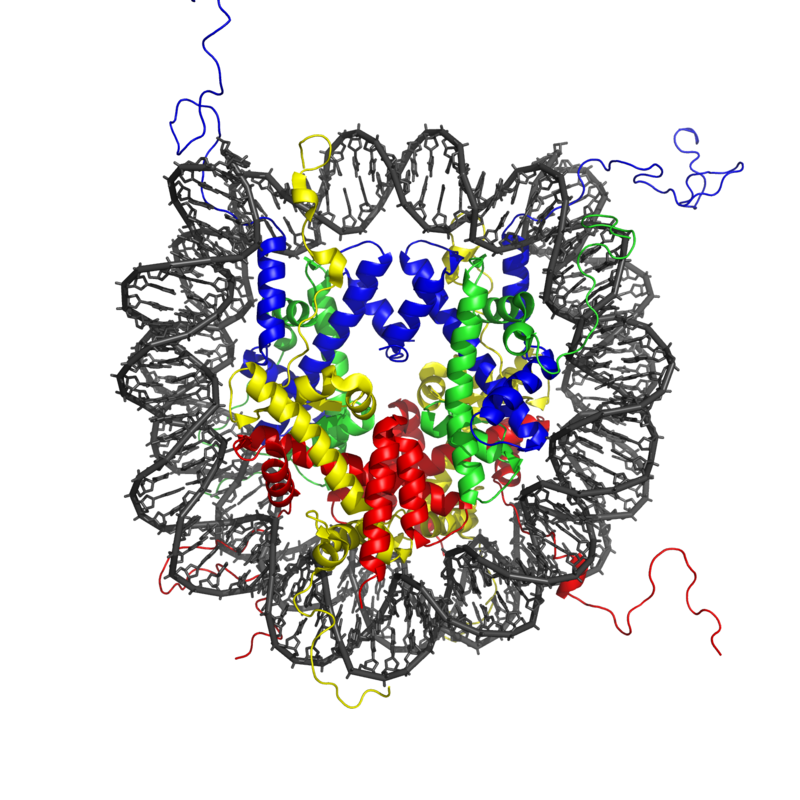
The modification of histone proteins comprises an important epigenetic mark related to gene expression. One of the most thoroughly studied modifications is histone acetylation, which is associated with gene activation and increased gene expression (Wade, Pruss, & Wolffe, 1997). Acetylation on histone tails is mediated by the opposing enzymatic activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Kuo & Allis, 1998). For example, acetylation of histone in gene regulatory regions by HAT enzymes is generally associated with DNA demethylation, gene activation, and increased gene expression (Hong, Schroth, Matthews, Yau, & Bradbury, 1993; Sealy & Chalkley, 1978). On the other hand, removal of the acetyl group (deacetylation) by HDAC enzymes is generally associated with DNA methylation, gene silencing, and decreased gene expression (Davie & Chadee, 1998). The relationship between patterns of histone modifications and gene activity provides evidence for the existence of a “histone code” for determining cell-specific gene expression programs (Jenuwein & Allis, 2001). Interestingly, recent research using animal models has demonstrated that histone modifications and DNA methylation of certain genes mediates the long-term behavioral effects of the level of care experienced during infancy.
Early childhood experience
The development of an individual is an active process of adaptation that occurs within a social and economic context. For example, the closeness or degree of positive attachment of the parent (typically mother)–infant bond and parental investment (including nutrient supply provided by the parent) that define early childhood experience also program the development of individual differences in stress responses in the brain, which then affect memory, attention, and emotion. In terms of evolution, this process provides the offspring with the ability to physiologically adjust gene expression profiles contributing to the organization and function of neural circuits and molecular pathways that support (1) biological defensive systems for survival (e.g., stress resilience), (2) reproductive success to promote establishment and persistence in the present environment, and (3) adequate parenting in the next generation (Bradshaw, 1965).
Parental investment and programming of stress responses in the offspring
The most comprehensive study to date of variations in parental investment and epigenetic inheritance in mammals is that of the maternally transmitted responses to stress in rats. In rat pups, maternal nurturing (licking and grooming) during the first week of life is associated with long-term programming of individual differences in stress responsiveness, emotionality, cognitive performance, and reproductive behavior (Caldji et al., 1998; Francis, Diorio, Liu, & Meaney, 1999; Liu et al., 1997; Myers, Brunelli, Shair, Squire, & Hofer, 1989; Stern, 1997). In adulthood, the offspring of mothers that exhibit increased levels of pup licking and grooming over the first week of life show increased expression of the glucocorticoid receptor in the hippocampus (a brain structure associated with stress responsivity as well as learning and memory) and a lower hormonal response to stress compared with adult animals reared by low licking and grooming mothers (Francis et al., 1999; Liu et al., 1997). Moreover, rat pups that received low levels of maternal licking and grooming during the first week of life showed decreased histone acetylation and increased DNA methylation of a neuron-specific promoter of the glucocorticoid receptor gene (Weaver et al., 2004). The expression of this gene is then reduced, the number of glucocorticoid receptors in the brain is decreased, and the animals show a higher hormonal response to stress throughout their life. The effects of maternal care on stress hormone responses and behaviour in the offspring can be eliminated in adulthood by pharmacological treatment (HDAC inhibitor trichostatin A, TSA) or dietary amino acid supplementation (methyl donor L-methionine), treatments that influence histone acetylation, DNA methylation, and expression of the glucocorticoid receptor gene (Weaver et al., 2004; Weaver et al., 2005). This series of experiments shows that histone acetylation and DNA methylation of the glucocorticoid receptor gene promoter is a necessary link in the process leading to the long-term physiological and behavioral sequelae of poor maternal care. This points to a possible molecular target for treatments that may reverse or ameliorate the traces of childhood maltreatment.

Several studies have attempted to determine to what extent the findings from model animals are transferable to humans. Examination of post-mortem brain tissue from healthy human subjects found that the human equivalent of the glucocorticoid receptor gene promoter (NR3C1 exon 1F promoter) is also unique to the individual (Turner, Pelascini, Macedo, & Muller, 2008). A similar study examining newborns showed that methylation of the glucocorticoid receptor gene promoter maybe an early epigenetic marker of maternal mood and risk of increased hormonal responses to stress in infants 3 months of age (Oberlander et al., 2008). Although further studies are required to examine the functional consequence of this DNA methylation, these findings are consistent with our studies in the neonate and adult offspring of low licking and grooming mothers that show increased DNA methylation of the promoter of the glucocorticoid receptor gene, decreased glucocorticoid receptor gene expression, and increased hormonal responses to stress (Weaver et al., 2004). Examination of brain tissue from suicide victims found that the human glucocorticoid receptor gene promoter is also more methylated in the brains of individuals who had experienced maltreatment during childhood (McGowan et al., 2009). These finding suggests that DNA methylation mediates the effects of early environment in both rodents and humans and points to the possibility of new therapeutic approaches stemming from translational epigenetic research. Indeed, similar processes at comparable epigenetic labile regions could explain why the adult offspring of high and low licking/grooming mothers exhibit widespread differences in hippocampal gene expression and cognitive function (Weaver, Meaney, & Szyf, 2006).
However, this type of research is limited by the inaccessibility of human brain samples. The translational potential of this finding would be greatly enhanced if the relevant epigenetic modification can be measured in an accessible tissue. Examination of blood samples from adult patients with bipolar disorder, who also retrospectively reported on their experiences of childhood abuse and neglect, found that the degree of DNA methylation of the human glucocorticoid receptor gene promoter was strongly positively related to the reported experience of childhood maltreatment decades earlier. For a relationship between a molecular measure and reported historical exposure, the effects size is extraordinarily large. This opens a range of new possibilities: given the large effect size and consistency of this association, measurement of the GR promoter methylation may effectively become a blood test measuring the physiological traces left on the genome by early experiences. Although this blood test cannot replace current methods of diagnosis, this unique and addition information adds to our knowledge of how disease may arise and be manifested throughout life. Near-future research will examine whether this measure adds value over and above simple reporting of early adversities when it comes to predicting important outcomes, such as response to treatment or suicide.
Child nutrition and the epigenome

The old adage “you are what you eat” might be true on more than just a physical level: The food you choose (and even what your parents and grandparents chose) is reflected in your own personal development and risk for disease in adult life (Wells, 2003). Nutrients can reverse or change DNA methylation and histone modifications, thereby modifying the expression of critical genes associated with physiologic and pathologic processes, including embryonic development, aging, and carcinogenesis. It appears that nutrients can influence the epigenome either by directly inhibiting enzymes that catalyze DNA methylation or histone modifications, or by altering the availability of substrates necessary for those enzymatic reactions. For example, rat mothers fed a diet low in methyl group donors during pregnancy produce offspring with reduced DNMT-1 expression, decreased DNA methylation, and increased histone acetylation at promoter regions of specific genes, including the glucocorticoid receptor, and increased gene expression in the liver of juvenile offspring (Lillycrop, Phillips, Jackson, Hanson, & Burdge, 2005) and adult offspring (Lillycrop et al., 2007). These data suggest that early life nutrition has the potential to influence epigenetic programming in the brain not only during early development but also in adult life, thereby modulating health throughout life. In this regard, nutritional epigenetics has been viewed as an attractive tool to prevent pediatric developmental diseases and cancer, as well as to delay aging-associated processes.
The best evidence relating to the impact of adverse environmental conditions development and health comes from studies of the children of women who were pregnant during two civilian famines of World War II: the Siege of Leningrad (1941–44) (Bateson, 2001) and the Dutch Hunger Winter (1944–1945) (Stanner et al., 1997). In the Netherlands famine, women who were previously well nourished were subjected to low caloric intake and associated environmental stressors. Women who endured the famine in the late stages of pregnancy gave birth to smaller babies (Lumey & Stein, 1997) and these children had an increased risk of insulin resistance later in life (Painter, Roseboom, & Bleker, 2005). In addition, offspring who were starved prenatally later experienced impaired glucose tolerance in adulthood, even when food was more abundant (Stanner et al., 1997). Famine exposure at various stages of gestation was associated with a wide range of risks such as increased obesity, higher rates of coronary heart disease, and lower birth weight (Lumey & Stein, 1997). Interestingly, when examined 60 years later, people exposed to famine prenatally showed reduced DNA methylation compared with their unexposed same-sex siblings (Heijmans et al., 2008).
Epigenetic regulation of learning and memory
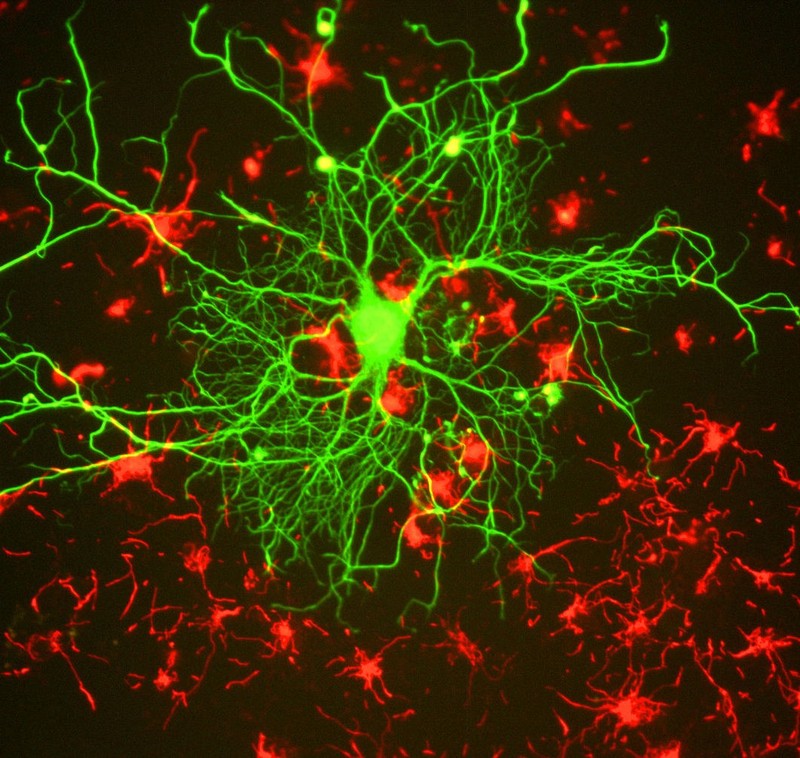
Memories are recollections of actual events stored within our brains. But how is our brain able to form and store these memories? Epigenetic mechanisms influence genomic activities in the brain to produce long-term changes in synaptic signaling, organization, and morphology, which in turn support learning and memory (Day & Sweatt, 2011).
Neuronal activity in the hippocampus of mice is associated with changes in DNA methylation (Guo et al., 2011), and disruption to genes encoding the DNA methylation machinery cause learning and memory impairments (Feng et al., 2010). DNA methylation has also been implicated in the maintenance of long-term memories, as pharmacological inhibition of DNA methylation and impaired memory (Day & Sweatt, 2011; Miller et al., 2010). These findings indicate the importance of DNA methylation in mediating synaptic plasticity and cognitive functions, both of which are disturbed in psychological illness.
Changes in histone modifications can also influence long-term memory formation by altering chromatin accessibility and the expression of genes relevant to learning and memory. Memory formation and the associated enhancements in synaptic transmission are accompanied by increases in histone acetylation (Guan et al., 2002) and alterations in histone methylation (Schaefer et al., 2009), which promote gene expression. Conversely, a neuronal increase in histone deacetylase activity, which promotes gene silencing, results in reduced synaptic plasticity and impairs memory (Guan et al., 2009). Pharmacological inhibition of histone deacetylases augments memory formation (Guan et al., 2009; Levenson et al., 2004), further suggesting that histone (de)acetylation regulates this process.
In humans genetic defects in genes encoding the DNA methylation and chromatin machinery exhibit profound effects on cognitive function and mental health (Jiang, Bressler, & Beaudet, 2004). The two best-characterized examples are Rett syndrome (Amir et al., 1999) and Rubinstein-Taybi syndrome (RTS) (Alarcon et al., 2004), which are profound intellectual disability disorders. Both MECP2 and CBP are highly expressed in neurons and are involved in regulating neural gene expression (Chen et al., 2003; Martinowich et al., 2003).
Rett syndrome patients have a mutation in their DNA sequence in a gene called MECP2. MECP2 plays many important roles within the cell: One of these roles is to read the DNA sequence, checking for DNA methylation, and to bind to areas that contain methylation, thereby preventing the wrong proteins from being present. Other roles for MECP2 include promoting the presence of particular, necessary, proteins, ensuring that DNA is packaged properly within the cell and assisting with the production of proteins. MECP2 function also influences gene expression that supports dendritic and synaptic development and hippocampus-dependent memory (Li, Zhong, Chau, Williams, & Chang, 2011; Skene et al., 2010). Mice with altered MECP2 expression exhibit genome-wide increases in histone acetylation, neuron cell death, increased anxiety, cognitive deficits, and social withdrawal (Shahbazian et al., 2002). These findings support a model in which DNA methylation and MECP2 constitute a cell-specific epigenetic mechanism for regulation of histone modification and gene expression, which may be disrupted in Rett syndrome.
RTS patients have a mutation in their DNA sequence in a gene called CBP. One of these roles of CBP is to bind to specific histones and promote histone acetylation, thereby promoting gene expression. Consistent with this function, RTS patients exhibit a genome-wide decrease in histone acetylation and cognitive dysfunction in adulthood (Kalkhoven et al., 2003). The learning and memory deficits are attributed to disrupted neural plasticity (Korzus, Rosenfeld, & Mayford, 2004). Similar to RTS in humans, mice with a mutation of CBP perform poorly in cognitive tasks and show decreased genome-wide histone acetylation (for review, see Josselyn, 2005). In the mouse brain CBP was found to act as an epigenetic switch to promote the birth of new neurons in the brain. Interestingly, this epigenetic mechanism is disrupted in the fetal brains of mice with a mutation of CBP, which, as pups, exhibit early behavioral deficits following removal and separation from their mother (Wang et al., 2010). These findings provide a novel mechanism whereby environmental cues, acting through histone modifying enzymes, can regulate epigenetic status and thereby directly promote neurogenesis, which regulates neurobehavioral development.
Together, these studies demonstrate that misregulation of epigenetic modifications and their regulatory enzymes is capable of orchestrating prominent deficits in neuronal plasticity and cognitive function. Knowledge from these studies may provide greater insight into other mental disorders such as depression and suicidal behaviors.
Epigenetic mechanisms in psychological disorders
Epigenome-wide studies have identified several dozen sites with DNA methylation alterations in genes involved in brain development and neurotransmitter pathways, which had previously been associated with mental illness (Mill et al., 2008). These disorders are complex and typically start at a young age and cause lifelong disability. Often, limited benefits from treatment make these diseases some of the most burdensome disorders for individuals, families, and society. It has become evident that the efforts to identify the primary causes of complex psychiatric disorders may significantly benefit from studies linking environmental effects with changes observed within the individual cells.
Epigenetic events that alter chromatin structure to regulate programs of gene expression have been associated with depression-related behavior and action of antidepressant medications, with increasing evidence for similar mechanisms occurring in post-mortem brains of depressed individuals. In mice, social avoidance resulted in decreased expression of hippocampal genes important in mediating depressive responses (Tsankova et al., 2006). Similarly, chronic social defeat stress was found to decrease expression of genes implicated in normal emotion processing (Lutter et al., 2008). Consistent with these findings, levels of histone markers of increased gene expression were down regulated in human post-mortem brain samples from individuals with a history of clinical depression (Covington et al., 2009).
Administration of antidepressants increased histone markers of increased gene expression and reversed the gene repression induced by defeat stress (Lee, Wynder, Schmidt, McCafferty, & Shiekhattar, 2006; Tsankova et al., 2006; Wilkinson et al., 2009). These results provide support for the use of HDAC inhibitors against depression. Accordingly, several HDAC inhibitors have been found to exert antidepressant effects by each modifying distinct cellular targets (Cassel et al., 2006; Schroeder, Lin, Crusio, & Akbarian, 2007).
There is also increasing evidence that aberrant gene expression resulting from altered epigenetic regulation is associated with the pathophysiology of suicide (McGowan et al., 2008; Poulter et al., 2008). Thus, it is tempting to speculate that there is an epigenetically determined reduced capacity for gene expression, which is required for learning and memory, in the brains of suicide victims.
Epigenetic strategy to understanding gene-environment interactions

While the cellular and molecular mechanisms that influence on physical and mental health have long been a central focus of neuroscience, only in recent years has attention turned to the epigenetic mechanisms behind the dynamic changes in gene expression responsible for normal cognitive function and increased risk for mental illness. The links between early environment and epigenetic modifications suggest a mechanism underlying gene-environment interactions. Early environmental adversity alone is not a sufficient cause of mental illness, because many individuals with a history of severe childhood maltreatment or trauma remain healthy. It is increasingly becoming evident that inherited differences in the segments of specific genes may moderate the effects of adversity and determine who is sensitive and who is resilient through a gene-environment interplay. Genes such as the glucocorticoid receptor appear to moderate the effects of childhood adversity on mental illness. Remarkably, epigenetic DNA modifications have been identified that may underlie the long-lasting effects of environment on biological functions. This new epigenetic research is pointing to a new strategy to understanding gene-environment interactions.
The next decade of research will show if this potential can be exploited in the development of new therapeutic options that may alter the traces that early environment leaves on the genome. However, as discussed in this module, the epigenome is not static and can be molded by developmental signals, environmental perturbations, and disease states, which present an experimental challenge in the search for epigenetic risk factors in psychological disorders (Rakyan, Down, Balding, & Beck, 2011). The sample size and epigenomic assay required is dependent on the number of tissues affected, as well as the type and distribution of epigenetic modifications. The combination of genetic association maps studies with epigenome-wide developmental studies may help identify novel molecular mechanisms to explain features of inheritance of personality traits and transform our understanding of the biological basis of psychology. Importantly, these epigenetic studies may lead to identification of novel therapeutic targets and enable the development of improved strategies for early diagnosis, prevention, and better treatment of psychological and behavioral disorders.
Outside Resources
Reference: The “Encyclopedia of DNA Elements” (ENCODE) project http://encodeproject.org/ENCODE/
Reference: THREADS – A new way to explore the ENCODE Project http://www.nature.com/encode/#/threads
Web: Explore, view, and download genome-wide maps of DNA and histone modifications from the NCBI Epigenomics Portal http://www.ncbi.nlm.nih.gov/epigenomics
Web: NOVA ScienceNOW – Introduction to Epigenetics http://www.pbs.org/wgbh/nova/genes
Web: The University of Utah’s Genetic Science Learning Center http://learn.genetics.utah.edu/content/epigenetics/
Discussion Questions
- Describe the physical state of the genome when genes are active and inactive.
- Often, the physical characteristics of genetically identical twins become increasingly different as they age, even at the molecular level. Explain why this is so (use the terms “environment” and “epigenome”).
- Name 3–4 environmental factors that influence the epigenome and describe their effects.
- The rat nurturing example shows us how parental behavior can shape the behavior of offspring on a biochemical level. Discuss how this relates to humans and include the personal and social implications.
- Explain how the food we eat affects gene expression.
- Can the diets of parents affect their offspring’s epigenome?
- Why is converging evidence the best kind of evidence in the study of brain function?
- If you were interested in whether a particular brain area was involved in a specific behavior, what neuroscience methods could you use?
- If you were interested in the precise time in which a particular brain process occurred, which neuroscience methods could you use?
Image Attributions
Figure 4.10: CC0 Public Domain, https://goo.gl/m25gce
Figure 4.11: M., https://goo.gl/VU5iJv, CC BY-NC-SA 2.0, https://goo.gl/Toc0ZF
Figure 4.12: Zephyris, https://goo.gl/gGrSQd, CC BY-SA 3.0, https://goo.gl/kB1Ogc
Figure 4.13: The White Ribbon Alliance, https://goo.gl/KgY6N5, CC BY-NC-SA 2.0, https://goo.gl/Toc0ZF
Figure 4.14: U.S. Department of Agriculture, https://goo.gl/tpyYzA, CC BY 2.0, https://goo.gl/BRvSA7
Figure 4.15: Gerry Shaw, https://goo.gl/JBqlY7, CC BY-SA 3.0, https://goo.gl/eLCn2O
Figure 4.16: Zephyris, https://goo.gl/6DBQ1g, CC BY-SA 3.0, https://goo.gl/eLCn2O
Figure 4.17: Steve White, CC0 Public Domain, https://goo.gl/m25gce
References
Adams, R. L., McKay, E. L., Craig, L. M., & Burdon, R. H. (1979). Mouse DNA methylase: methylation of native DNA. Biochimica et Biophysica Acta, 561(2), 345–357.
Alarcon, J. M., Malleret, G., Touzani, K., Vronskaya, S., Ishii, S., Kandel, E. R., & Barco, A. (2004). Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron, 42(6), 947–959. doi: 10.1016/j.neuron.2004.05.021, S0896627304003022 [pii]
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., & Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics, 23(2), 185–188.
Bateson, P. (2001). Fetal experience and good adult design. International Journal of Epidemiology, 30(5), 928–934.
Bradshaw, A.D. (1965). Evolutionary significance of phenotypic plasticity in plants. Advances in Genetics, 13, 115–155.
Caldji, C., Tannenbaum, B., Sharma, S., Francis, D., Plotsky, P. M., & Meaney, M. J. (1998). Maternal care during infancy regulates the development of neural systems mediating the expression of fearfulness in the rat. Proceedings of the National Academy of Sciences U S A, 95(9), 5335–5340.
Cassel, S., Carouge, D., Gensburger, C., Anglard, P., Burgun, C., Dietrich, J. B., . . . Zwiller, J. (2006). Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Molecular Pharmacology, 70(2), 487–492. doi: 10.1124/mol.106.022301
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., . . . Greenberg, M. E. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science, 302(5646), 885–889.
Covington, H. E., 3rd, Maze, I., LaPlant, Q. C., Vialou, V. F., Ohnishi, Y. N., Berton, O., . . . Nestler, E. J. (2009). Antidepressant actions of histone deacetylase inhibitors. Journal of Neuroscience, 29(37), 11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009
Davie, J. R., & Chadee, D. N. (1998). Regulation and regulatory parameters of histone modifications. *Journal of Cellular Biochemistry Suppl, 30–31 *, 203–213.
Day, J. J., & Sweatt, J. D. (2011). Epigenetic mechanisms in cognition. Neuron, 70(5), 813–829. doi: 10.1016/j.neuron.2011.05.019
Dick, D. M., Riley, B., & Kendler, K. S. (2010). Nature and nurture in neuropsychiatric genetics: where do we stand? Dialogues in Clinical Neuroscience, 12(1), 7–23.
Dolinoy, D. C., Weidman, J. R., & Jirtle, R. L. (2007). Epigenetic gene regulation: linking early developmental environment to adult disease. Reproductive Toxicology, 23(3), 297–307. doi: S0890-6238(06)00197-3 [pii], 10.1016/j.reprotox.2006.08.012
Feng, J., Chang, H., Li, E., & Fan, G. (2005). Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. Journal of Neuroscience Research, 79(6), 734–746. doi: 10.1002/jnr.20404
Feng, J., Zhou, Y., Campbell, S. L., Le, T., Li, E., Sweatt, J. D., . . . Fan, G. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nature Neuroscience, 13(4), 423–430. doi: 10.1038/nn.2514
Francis, D., Diorio, J., Liu, D., & Meaney, M. J. (1999). Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science, 286(5442), 1155–1158.
Gershon, E. S., Alliey-Rodriguez, N., & Liu, C. (2011). After GWAS: searching for genetic risk for schizophrenia and bipolar disorder. American Journal of Psychiatry, 168(3), 253–256. doi: 10.1176/appi.ajp.2010.10091340
Goll, M. G., & Bestor, T. H. (2005). Eukaryotic cytosine methyltransferases. Annual Review of Biochemistry, 74, 481–514. doi: 10.1146/annurev.biochem.74.010904.153721
Goto, K., Numata, M., Komura, J. I., Ono, T., Bestor, T. H., & Kondo, H. (1994). Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation, 56(1–2), 39–44.
Gregg, C., Zhang, J., Weissbourd, B., Luo, S., Schroth, G. P., Haig, D., & Dulac, C. (2010). High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science, 329(5992), 643–648. doi: 10.1126/science.1190830
Guan, J. S., Haggarty, S. J., Giacometti, E., Dannenberg, J. H., Joseph, N., Gao, J., . . . Tsai, L. H. (2009). HDAC2 negatively regulates memory formation and synaptic plasticity. Nature, 459(7243), 55-60. doi: 10.1038/nature07925
Guan, Z., Giustetto, M., Lomvardas, S., Kim, J. H., Miniaci, M. C., Schwartz, J. H., . . . Kandel, E. R. (2002). Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell, 111(4), 483–493.
Guo, J. U., Ma, D. K., Mo, H., Ball, M. P., Jang, M. H., Bonaguidi, M. A., . . . Song, H. (2011). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nature Neuroscience, 14(10), 1345–1351. doi: 10.1038/nn.2900
Heijmans, B. T., Tobi, E. W., Stein, A. D., Putter, H., Blauw, G. J., Susser, E. S., . . . Lumey, L. H. (2008). Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences U S A, 105(44), 17046–17049. doi: 0806560105 [pii]10.1073/pnas.0806560105
Hong, L., Schroth, G. P., Matthews, H. R., Yau, P., & Bradbury, E. M. (1993). Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. Journal of Biological Chemistry, 268(1), 305–314.
Jenuwein, T., & Allis, C. D. (2001). Translating the histone code. Science, 293(5532), 1074–1080. doi: 10.1126/Science.1063127293/5532/1074 [pii]
Jiang, Y. H., Bressler, J., & Beaudet, A. L. (2004). Epigenetics and human disease. Annual Review of Genomics and Human Genetics, 5, 479–510. doi: 10.1146/annurev.genom.5.061903.180014
Josselyn, S. A. (2005). What’s right with my mouse model? New insights into the molecular and cellular basis of cognition from mouse models of Rubinstein-Taybi Syndrome. Learning & Memory, 12(2), 80–83. doi: 12/2/80 [pii]10.1101/lm.93505
Kadonaga, J. T. (1998). Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell, 92(3), 307–313.
Kalkhoven, E., Roelfsema, J. H., Teunissen, H., den Boer, A., Ariyurek, Y., Zantema, A., . . . Peters, D. J. (2003). Loss of CBP acetyltransferase activity by PHD finger mutations in Rubinstein-Taybi syndrome. Human Molecular Genetics, 12(4), 441–450.
Korzus, E., Rosenfeld, M. G., & Mayford, M. (2004). CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron, 42(6), 961–972. doi: 10.1016/j.neuron.2004.06.002S0896627304003526 [pii]
Kuo, M. H., & Allis, C. D. (1998). Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays, 20(8), 615–626. doi: 10.1002/(SICI)1521–1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H [pii] 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H
Law, J. A., & Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics, 11(3), 204–220. doi: nrg2719 [pii]10.1038/nrg2719
Lee, M. G., Wynder, C., Schmidt, D. M., McCafferty, D. G., & Shiekhattar, R. (2006). Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chemistry & Biology, 13(6), 563–567. doi: 10.1016/j.chembiol.2006.05.004
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., & Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. Journal of Biological Chemistry, 279(39), 40545–40559.
Li, H., Zhong, X., Chau, K. F., Williams, E. C., & Chang, Q. (2011). Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nature Neuroscience, 14(8), 1001–1008. doi: 10.1038/nn.2866
Lillycrop, K. A., Phillips, E. S., Jackson, A. A., Hanson, M. A., & Burdge, G. C. (2005). Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. Journal of Nutrition, 135(6), 1382–1386. doi: 135/6/1382 [pii]
Lillycrop, K. A., Slater-Jefferies, J. L., Hanson, M. A., Godfrey, K. M., Jackson, A. A., & Burdge, G. C. (2007). Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. British Journal of Nutrition, 97(6), 1064–1073. doi: S000711450769196X [pii]10.1017/S000711450769196X
Liu, D., Diorio, J., Tannenbaum, B., Caldji, C., Francis, D., Freedman, A., . . . Meaney, M. J. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic- pituitary-adrenal responses to stress [see comments]. Science, 277(5332), 1659–1662.
Lumey, L. H., & Stein, A. D. (1997). Offspring birth weights after maternal intrauterine undernutrition: a comparison within sibships. American Journal of Epidemiology, 146(10), 810–819.
Lutter, M., Krishnan, V., Russo, S. J., Jung, S., McClung, C. A., & Nestler, E. J. (2008). Orexin signaling mediates the antidepressant-like effect of calorie restriction. Journal of Neuroscience, 28(12), 3071–3075. doi: 10.1523/JNEUROSCI.5584-07.2008
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., . . . Sun, Y. E. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science, 302(5646), 890–893.
McGowan, P. O., Sasaki, A., D’Alessio, A. C., Dymov, S., Labonte, B., Szyf, M., . . . Meaney, M. J. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience, 12(3), 342–348. doi: nn.2270 [pii]10.1038/nn.2270
McGowan, P. O., Sasaki, A., Huang, T. C., Unterberger, A., Suderman, M., Ernst, C., . . . Szyf, M. (2008). Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS ONE, 3(5), e2085. doi: 10.1371/journal.pone.0002085
Mill, J., Tang, T., Kaminsky, Z., Khare, T., Yazdanpanah, S., Bouchard, L., . . . Petronis, A. (2008). Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. American Journal of Human Genetics, 82(3), 696–711. doi: 10.1016/j.ajhg.2008.01.008
Miller, C. A., Gavin, C. F., White, J. A., Parrish, R. R., Honasoge, A., Yancey, C. R., . . . Sweatt, J. D. (2010). Cortical DNA methylation maintains remote memory. Nature Neuroscience, 13(6), 664–666. doi: 10.1038/nn.2560
Myers, M. M., Brunelli, S. A., Shair, H. N., Squire, J. M., & Hofer, M. A. (1989). Relationships between maternal behavior of SHR and WKY dams and adult blood pressures of cross-fostered F1 pups. Developmental Psychobiology, 22(1), 55–67.
Oberlander, T. F., Weinberg, J., Papsdorf, M., Grunau, R., Misri, S., & Devlin, A. M. (2008). Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics, 3(2), 97–106. doi: 6034 [pii]
Ooi, S. K., O’Donnell, A. H., & Bestor, T. H. (2009). Mammalian cytosine methylation at a glance. Journal of Cell Science, 122(Pt 16), 2787–2791. doi: 122/16/2787 [pii]10.1242/jcs.015123
Painter, R. C., Roseboom, T. J., & Bleker, O. P. (2005). Prenatal exposure to the Dutch famine and disease in later life: an overview. Reproductive Toxicology, 20(3), 345–352. doi: S0890-6238(05)00088-2 [pii]10.1016/j. Reproductive Toxicology.2005.04.005
Petronis, A. (2010). Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature, 465(7299), 721–727. doi: 10.1038/nature09230
Poulter, M. O., Du, L., Weaver, I. C., Palkovits, M., Faludi, G., Merali, Z., . . . Anisman, H. (2008). GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biological Psychiatry, 64(8), 645–652. doi: 10.1016/j.biopsych.2008.05.028
Rakyan, V. K., Down, T. A., Balding, D. J., & Beck, S. (2011). Epigenome-wide association studies for common human diseases. Nature Reviews Genetics, 12(8), 529–541. doi: 10.1038/nrg3000
Razin, A. (1998). CpG methylation, chromatin structure and gene silencing-a three-way connection. European Molecular Biology Organization, 17(17), 4905–4908.
Schaefer, A., Sampath, S. C., Intrator, A., Min, A., Gertler, T. S., Surmeier, D. J., . . . Greengard, P. (2009). Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron, 64(5), 678–691. doi: 10.1016/j.neuron.2009.11.019
Schroeder, F. A., Lin, C. L., Crusio, W. E., & Akbarian, S. (2007). Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biological Psychiatry, 62(1), 55-64. doi: 10.1016/j.biopsych.2006.06.036
Sealy, L., & Chalkley, R. (1978). DNA associated with hyperacetylated histone is preferentially digested by DNase I. Nucleic Acids Research, 5(6), 1863–1876.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., . . . Zoghbi, H. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron, 35(2), 243–254.
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R., James, K. D., Turner, D. J., . . . Bird, A. P. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Molecular Cell, 37(4), 457–468. doi: 10.1016/j.molcel.2010.01.030
Stanner, S. A., Bulmer, K., Andres, C., Lantseva, O. E., Borodina, V., Poteen, V. V., & Yudkin, J. S. (1997). Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. British Medical Journal, 315(7119), 1342–1348.
Stern, J. M. (1997). Offspring-induced nurturance: animal-human parallels. Developmental Psychobiololgy, 31(1), 19–37.
Sutter, D., Doerfler, W., 1980. Methylation of integrated adenovirus type 12 DNA sequences in transformed cells is inversely correlated with viral gene expression. Proceedings of the National Academy of Sciences U S A. 77, 253–256.
Suzuki, M. M., & Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics, 9(6), 465–476. doi: nrg2341 [pii]10.1038/nrg2341
Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., & Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nature Neuroscience. 9(4): 519–525. doi:10.1038/nn1659
Turner, J. D., Pelascini, L. P., Macedo, J. A., & Muller, C. P. (2008). Highly individual methylation patterns of alternative glucocorticoid receptor promoters suggest individualized epigenetic regulatory mechanisms. Nucleic Acids Research, 36(22), 7207–7218. doi: gkn897 [pii] 10.1093/nar/gkn897
Vardimon, L., Kressmann, A., Cedar, H., Maechler, M., Doerfler, W., 1982. Expression of a cloned adenovirus gene is inhibited by in vitro methylation. Proceedings of the National Academy of Sciences U S A. 79, 1073–1077.
Waddington, C. H. (1942). Epigenotype. Endeavour(1), 18–21.
Wade, P. A., Pruss, D., & Wolffe, A. P. (1997). Histone acetylation: chromatin in action. Trends in Biochemical Sciences, 22(4), 128–132. doi: S0968000497010165 [pii]
Wang, J., Weaver, I. C., Gauthier-Fisher, A., Wang, H., He, L., Yeomans, J., . . . Miller, F. D. (2010). CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Developmental Cell, 18(1), 114–125. doi: 10.1016/j.devcel.2009.10.023
Weaver, I. C., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., . . . Meaney, M. J. (2004). Epigenetic programming by maternal behavior. Nature Neuroscience, 7(8), 847–854. doi: 10.1038/nn1276
Weaver, I. C., Champagne, F. A., Brown, S. E., Dymov, S., Sharma, S., Meaney, M. J., & Szyf, M. (2005). Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. Journal of Neuroscience, 25(47), 11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005
Weaver, I. C., Meaney, M. J., & Szyf, M. (2006). Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proceedings of the National Academy of Sciences U S A, 103(9), 3480–3485. doi: 10.1073/pnas.0507526103
Wells, J. C. (2003). The thrifty phenotype hypothesis: thrifty offspring or thrifty mother? Journal of Theoretical Biology, 221(1), 143–161.
Wilkinson, M. B., Xiao, G., Kumar, A., LaPlant, Q., Renthal, W., Sikder, D., . . . Nestler, E. J. (2009). Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. Journal of Neuroscience, 29(24), 7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009
Wolffe, A. P., & Matzke, M. A. (1999). Epigenetics: regulation through repression. Science, 286(5439), 481–486.
