Digging Deeper
14 Soil Mineralogy
Jim Warren and Graeme Spiers
LEARNING OUTCOMES
On completion of this chapter, students should be able to:
- Identify common mineral phases found in soil
- Explain the difference between primary and secondary minerals
- Describe the influence of continental glaciation on mineral distributions in Canadian soils
- Describe the relationship of between particle size and soil minerals
- Define isomorphous substitution and the basis for cation exchange capacity
- Explain differences in the phyllosilicate composition of soils between eastern and western Canada
INTRODUCTION
Soil mineralogy is “concerned with the inorganic minerals found in the pedosphere and to the depth of weathering” (Finkl, 1983). Although soil mineralogy draws heavily on the disciplines of mineralogy, geology, inorganic chemistry and crystallography—which are scientific disciplines unto themselves—the reader is not required to have much prior knowledge in these areas to understand the information presented herein. The general approach taken here is based on the textural fractions of soil (sand, silt and clay) and the mineral components contained therein.
Importance of the Soil Mineral Fraction
Most soils are composed of inorganic mineral materials that comprise the overwhelming bulk of a soil. Well-structured soils are composed of about 50% solids of which most is mineral materials (Figure 14.1), with organic soils (>30% organic matter) being the obvious exception.
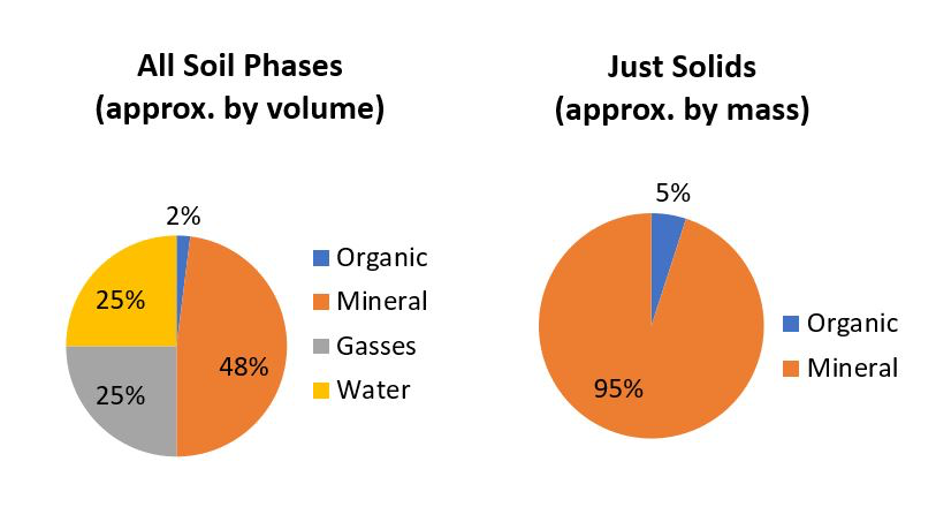
There are about 4,500 to 5,000 named minerals identified on this planet (MSA, 1997–2020; MSA, 2004–2020; Mindat, 2020), with most being either rare or found in isolated pockets (e.g., economic mineral deposits) deep in the Earth’s crust. Minerals are naturally occurring inorganic crystals with a definable range of chemical composition. For example, by far the most common terrestrial mineral found in Canadian soils is α-quartz (α-SiO2); pronounced “alpha-quartz” where alpha refers to the crystal form. Rock fragments are also found in soil but these differ from minerals in that rocks are “mineral composites” made up of physical mixtures of several individual minerals. For example, granite is an igneous rock making up the vast majority of the Canadian Shield. Granite is commonly composed of a mixture of four types of minerals: α-quartz, feldspars, amphiboles and micas which, due to the grinding and mixing actions in the glaciers, are also common minerals found in our soils. Rock fragments are only found among the larger soil particles (i.e., sand-size and larger) because as rocks are broken down into increasingly smaller fragments, their physical breakdown typically occurs along boundaries between adjacent minerals, which are the weakest points within rocks, leaving the individual discrete minerals as the smaller fragments.
Most minerals found in soil are of the common variety, meaning they are ubiquitous and present in large quantities usually with little or no economic value. The types of minerals in soil represent only a handful (<100) compared to all the known minerals; however, their behaviour in soil as they interact with the soil solution (soil water), the soil atmosphere and soil organic matter have a huge bearing on the chemical characteristics and nature of our soils.
Soil minerals are the natural source of plant nutrients that are released slowly with time during chemical weathering. All plants require a minimum of 17 nutrient elements to complete their life cycle (see Chapter 7). With the exception of C, H, and O, which plants obtain from air and water, plants derive the remaining 14 elements (N, P, K, Ca, S, Mg, Fe, B, Cl, Mn, Zn, Cu, Mo, and Ni) mainly from the soil itself and through the addition of fertilizers, manures, and other amendments (Parikh and James, 2012; Singh and Schulze, 2015). An additional four elements (Na, Si, V, and Se) are essential to the growth of some plants (Havlin et al., 2005). Soil minerals also contribute to a soil’s ability to retain plant nutrients through the processes of cation and anion exchanges.
SOIL PARENT MATERIALS
Canadian soils are very young compared to those in the tropics, which have never been reworked by glaciers (see Chapter 2). Canada contains more glaciated terrain than any other country in the world (Rutter, 2015) and almost all Canadian soils are developed on Late Wisconsin glacial sediments that were laid down between about 5,000 to 18,000 years ago. The only exceptions are some small areas such as the Cypress Hills region of southern Alberta and Saskatchewan; non-glaciated areas of the northern Yukon and small parts of the western Northwest Territories which were not covered with ice during Late Wisconsin glaciation (see Chapter 2). By comparison soils such as those in the southern United States, Australia, Africa, and South America are several million years old. Therefore, Canadian soils are composed of inherited minerals derived mostly from the underlying bedrock and through transport and deposition by glacial ice during ice advance or by glacial melt waters and wind during deglaciation.
PRIMARY vs. SECONDARY MINERALS
The Earth’s crust may be described in the very broad context as being composed of two main lithologies: the oceanic crust composed mostly of mafic and ultramafic (Fe and Mg rich) rocks; and the continental crust which contains more granitic, metamorphic and sedimentary rocks. Since soils are located in terrestrial areas they are developed primarily from the continental crust rocks, which in the Canadian context is about 85% igneous and metamorphic rocks and 15% sedimentary rocks.
Minerals derived directly from geological and geochemical processes in the Earth’s crust are referred to as primary minerals. Examination of mineral surfaces with a microscope may reveal some changes due to biogeochemical weathering (Spiers, et al., 1989), but most primary minerals in Canadian soils have been only minimally altered by chemical actions in the last 5,000–18,000 years.
Once brought to the Earth’s surface most primary minerals are not chemically stable. Although they persist in our soils, given enough geological time they will eventually succumb to the biogeochemical processes of the environment and gradually release their elemental components to the environment. Primary minerals weather and dissolve at different rates. Generally, minerals containing Na, and K dissolve faster than those containing Ca and Mg, which in turn dissolve faster than minerals composed of Si, Al, and Fe. Iron is a slightly special case as it is the only major element with two valences (Fe2+ and Fe3+) with solubility differing greatly depending on the chemical environment. Under reducing (absence of oxygen) conditions, Fe tends to be soluble and present as the Fe2+ ion. Under oxidizing conditions, Fe2+ reacts with oxygen (oxidized) to form Fe3+ ions which in turn form insoluble oxyhydroxide minerals (effectively the composition of rust). Other minor elements (e.g., Mn, As, U, etc.) are transformed similarly. The result is that as weathering proceeds soils become generally depleted in Na, K, Ca and Mg which are leached from the soils while becoming enriched in Si, Al, and Fe3+ (see Figure 14.2). This also explains why the oceans contain elevated concentrations of soluble salts (Na, K, Cl and SO4). Carbonate minerals (principally CaCO3) are formed or dissolved depending on the pH of their bearing solution. They precipitate under alkaline conditions and are dissolved under acid conditions. Atmospheric CO2 reacts with the water to produce a weak acid (H2CO3) that dissolves carbonate (and other rock types). Deeper groundwater tends to be alkaline thus favouring precipitation of carbonate typically found deeper in soil profiles (C horizons).
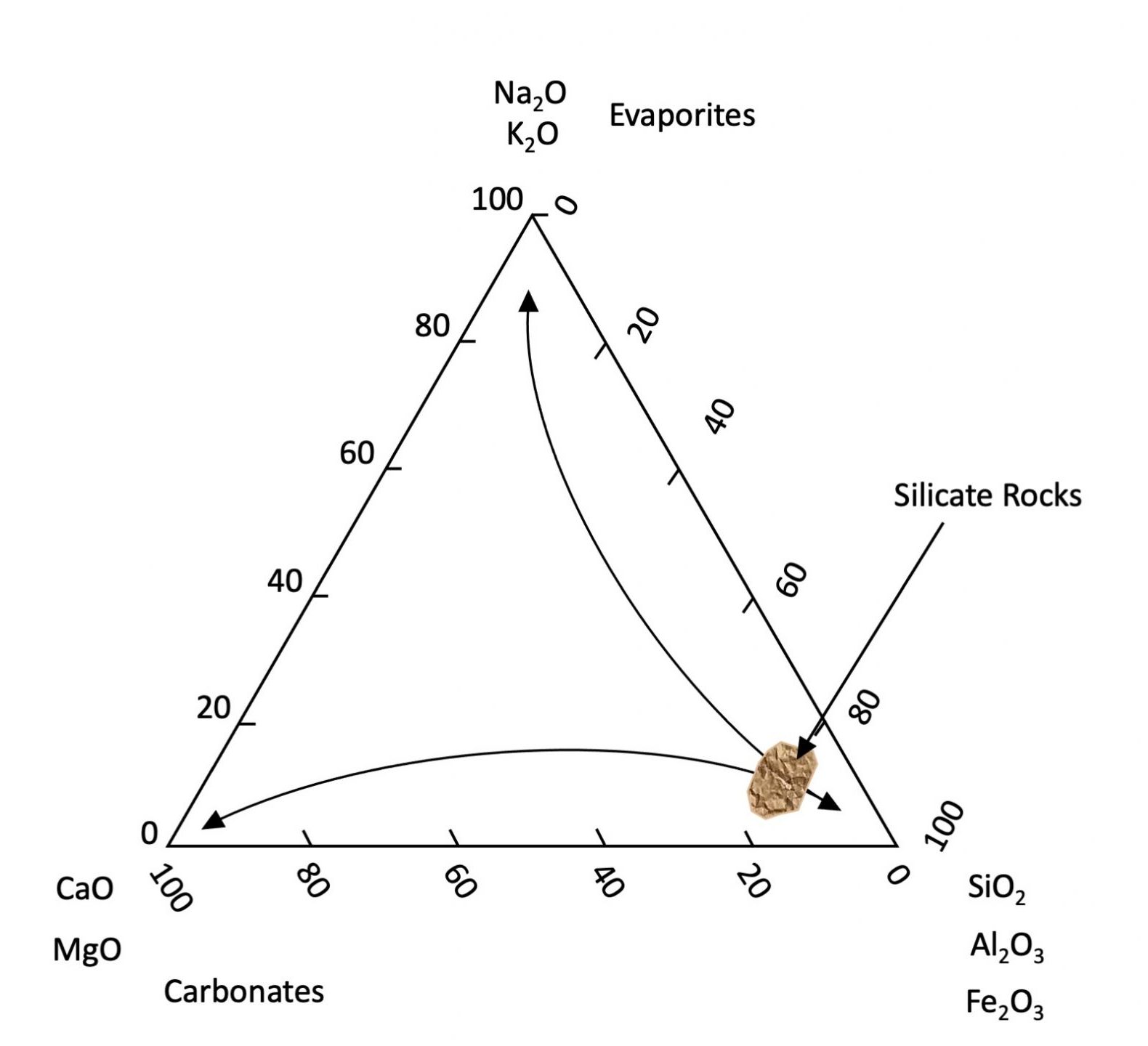
While primary minerals form by geological processes, secondary minerals are a by-product of chemical weathering formed either through the partial chemical alteration of primary minerals or through chemical precipitation from the reaction of components dissolved from other minerals. Most secondary minerals are hydrated aluminosilicates or Fe and Al oxyhydroxides that are stable in soils. Most hydrated aluminosilicates are classified as phyllosilicates or clay minerals which are the most abundant secondary minerals in Canadian soils. These are very fine-grained sheet-like particles with a very high surface area to mass ratio referred to as specific surface area (SSA). Since surface area is inversely related to particle diameter, the clay-size fraction accounts by far for the largest mineral portion of the available surface area associated with soil minerals (Chapter 5, Table 5.1).
Clay minerals are also the most chemically reactive inorganic phases in soils and both the amount and type of clay mineral influence many soil properties including:
- porosity and pore size;
- swelling behaviour;
- compressibility and compactability;
- sorption capacity;
- exchange capacity; and,
- hydraulic conductivity.
Can You Dig It!
Although secondary minerals are formed and are stable in soils at the surface of the Earth, the vast majority secondary minerals found in Canadian soils were formed in the distant past, prior to Late Wisconsin glaciation. Most soil minerals re-deposited during the last glaciation were derived from surface sediments formed and deposited, likely repeatedly, during the millennia prior to the last glaciation. Although secondary minerals continue to form in our soils in response to chemical weathering, the total amounts produced since the last glacial event are very small compared to the amounts re-deposited during many repeated glaciations in the past.
MINERALS vs PARTICLE SIZE
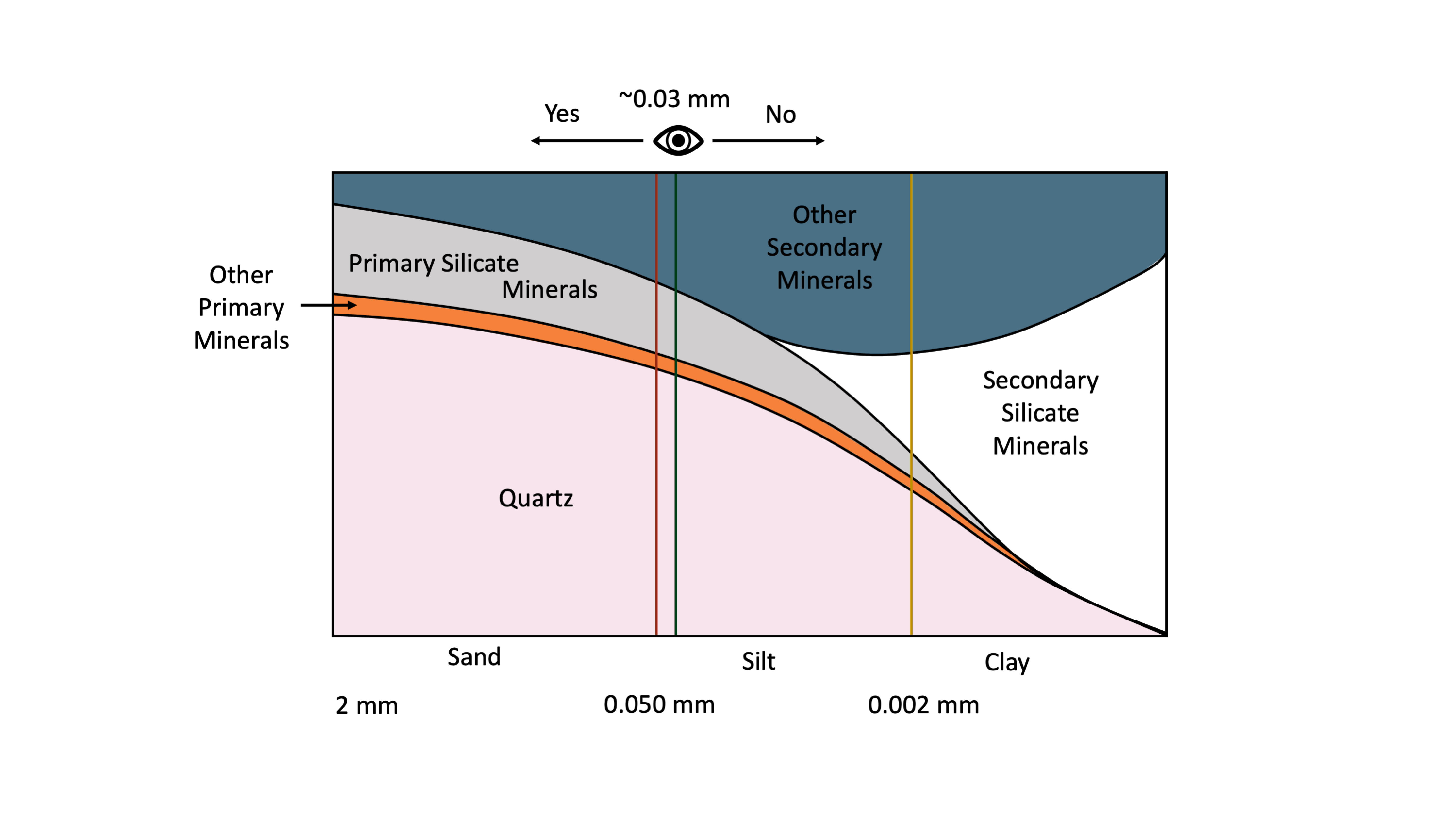
The average person with 20:20 vision can resolve individual particles as small as about 30 μm (0.03 mm) with the unaided eye. This is the approximate division (0.05 mm) between sand and silt-size soil particles (Figure 14.3). By comparison, the thickness of human hair is in the range of 0.1 to 0.04 mm. This means that the average individual with reasonably good eye-sight can easily distinguish individual sand-size particles with the naked eye (without magnification) but cannot differentiate between most individual silt particles and definitely cannot resolve clay-size particles. Examination of silt-size particles requires magnification through use of a light microscope while clay-size particles can only be resolved using electron microscopy (Smart and Tovey, 1981) or atomic force microscopy (Liu, 2003).
The distribution of soil minerals varies with particle size (Figure 14.3). Based on grain size fractions as defined in the Canadian System of Soil Classification (SCWG 1998), the sand-size fraction (particles with diameter in the range of 2-0.050 mm) are dominated by primary minerals. The primary minerals are mainly quartz, feldspars and other primary silicate minerals while the clay-size fraction (<0.002 mm diameter) is dominated by secondary phyllosilicates and iron and aluminum oxyhydroxides. The silt-size fraction (0.05-0.002 mm diameter) is intermediate in size between sand and clay and typically contains a mixture of both primary and secondary minerals.
Elemental Abundance and Mineral Structures
There are 91 naturally occurring elements, all of which are found at some concentration in soil. Table 14.1 provides a list of 70 elements in order of decreasing abundance in crustal rocks and in soils. Most elements substitute for one another within the crystal structure of minerals depending on their size and valence. This will be discussed in more detail later in this chapter. Depending on solubility and resistance to chemical weathering, the content of some elements contained in soil minerals increase relative to crustal abundance (e.g., Si and O) while the concentration of most elements decreases as they are released to the environment (compare crustal elemental abundance with soil abundance; Table 14.1).
Table 14.1. Total content (mg kg-1) of selected elements in order of abundance in continental crust rocks and in soils (Adapted from Bowen, 1979)
| Element | Crust | Soil | Element | Crust | Soil | |
|---|---|---|---|---|---|---|
| O | 464000 | 490000 | B | 10 | 38 | |
| Si | 281500 | 330000 | Th | 9.6 | 14 | |
| Al | 81650 | 71000 | Sm | 6 | 6 | |
| Fe | 53000 | 32000 | Gd | 5.4 | 3.5 | |
| Ca | 38650 | 15000 | Cs | 3 | 3 | |
| Na | 26150 | 10900 | Dy | 3 | 5.7 | |
| K | 23950 | 18300 | Yb | 3 | 3.9 | |
| Mg | 21950 | 8300 | Hf | 3 | 7.7 | |
| Ti | 5050 | 5100 | Be | 2.8 | 1.5 | |
| P | 1050 | 800 | Er | 2.8 | 3 | |
| Mn | 950 | 760 | U | 2.7 | 2.2 | |
| F | 625 | 270 | Br | 2.5 | 43 | |
| Ba | 425 | 568 | Sn | 2 | 5.8 | |
| Sr | 375 | 278 | Ta | 2 | 1.2 | |
| S | 260 | 433 | As | 1.8 | 11 | |
| Zr | 165 | 345 | Ge | 1.5 | 3 | |
| V | 135 | 108 | Mo | 1.5 | 1.9 | |
| Cl | 130 | 485 | W | 1.5 | 1.1 | |
| Cr | 100 | 84 | Eu | 1.2 | 1.3 | |
| Rb | 90 | 120 | Ho | 1.2 | 0.8 | |
| Ni | 75 | 34 | Tb | 0.9 | 0.85 | |
| Zn | 70 | 60 | I | 0.5 | 7.1 | |
| Ce | 60 | 84 | Lu | 0.5 | 0.46 | |
| Cu | 55 | 26 | Tm | 0.48 | 0.62 | |
| Y | 33 | 28 | Tl | 0.45 | 0.25 | |
| La | 30 | 41 | Cd | 0.2 | 0.6 | |
| Nd | 28 | 44 | Sb | 0.2 | 1.7 | |
| Co | 25 | 12 | Bi | 0.17 | 0.5 | |
| Sc | 22 | 10 | In | 0.1 | 1 | |
| Li | 20 | 31 | Hg | 0.08 | 0.1 | |
| Nb | 20 | 14 | Ag | 0.07 | 0.05 | |
| Ga | 15 | 21 | Se | 0.05 | 0.4 | |
| Pb | 12 | 29 | Pt | 0.001 | 0.001 | |
| Pr | 11 | 6.5 | Au | 0.001 | 0.001 | |
| Note: Does not include hydrogen, carbon, nitrogen, noble gasses, or elements found in ultra-trace (<0.001 mg kg-1) amounts. See https://earthref.org/GERMRD/10/ for the most updated version of this table. See Spiers et al. (1989a) for selected elements in Alberta soils. | ||||||
Closer examination of elemental abundances indicates that the eight most abundant elements (O, Si, Al, Fe, Ca, Na, K and Mg) account for almost the entire mass (99%) of the Earth’s crust (Table 14.2). Considering the atomic radii further indicates that almost 85% of the Earth’s crust by volume is composed of oxygen as oxides. Specifically oxide based minerals (O2-) form the fundamental framework of most minerals in the Earth’s crust with the major cations (Si, Al, Fe, Ca, Na, K, and Mg) fitting between the oxygen atoms on the atomic level.
Table 14.2. Radius of O2- and major cations, crustal abundance, radius ratios and predicted coordination numbers
| Element | Atomic Radius (nm) | Abundance by Mass (%) | Abundance by Volume (%) | Radius Ratio | Predicted coordination number (CN) |
|---|---|---|---|---|---|
| O2- | 0.14 | 0.464 | 0.845 | na* | na* |
| Si4+ | 0.04 | 0.282 | 0.012 | 0.29 | 4 |
| Al3+ | 0.054 | 0.0817 | 0.0083 | 0.38 | 4 |
| Fe3+ | 0.065 | 0.053 | 0.0094 | 0.46 | 6 |
| Fe2+ | 0.078 | Incl.** | Incl.** | 0.56 | 6 |
| Ca2+ | 0.112 | 0.0387 | 0.0361 | 0.80 | 8 |
| Na+ | 0.097 | 0.0262 | 0.0285 | 0.69 | 6 |
| K+ | 0.151 | 0.022 | 0.0548 | 1.08 | 12 |
| Mg2+ | 0.072 | 0.024 | 0.0054 | 0.51 | 6 |
| Total | 0.9909 | 1 | |||
| Note: *na = not applicable. **: abundance of Fe2+ included with Fe3+ | |||||
The crystal structure of minerals at the atomic level may be easily visualized if we assume that oxygen and the other metal ions act as rigid spheres with fixed radii. Although not strictly true, this is a useful conceptual to visualize crystal structures. The geometry of these rigid spheres is governed by Pauling’s Rules (Pauling 1929) where the first rule states that the distance between a cation and anion is the sum of their radii. Ions in a crystal structure tend to gather as many oppositely charged ions around itself as size permits. The number of O2- ions that may surround a central cation is called the coordination number (CN). Large cations (e.g. K+) have high coordination numbers because of their large volumes while small cations (e.g., Si4+) have low coordination numbers (Figure 14.4). Also note that those cations with large atomic radii and low valence (K+ and Na+) are the most mobile while those with smaller radii and high charge (Si4+, Al3+ and Fe 3+) are far less mobile.

Since ions tend to surround themselves with as many oppositely charged ions as possible, predicting coordination numbers requires using only the relative sizes of the ions expressed as the radius ratio:
(1) 
Table 14.3 provides ranges of radius ratios correspond to common coordination numbers and the corresponding structural units. Some overlap and uncertainty arise because the assumption of treating ions as rigid spheres with constant radii is not strictly true. With this in mind, ions are typically interchangeable if their size does not differ by more than about 15%, and their valences do not differ by more than one unit. In reality, ions are not spherical and their radii change slightly with different coordination numbers. As a result, some elements have more than one common coordination geometry. For example, aluminum is commonly found in both tetrahedral (CN=4) and octahedral (CN=6) units. Substitution of cations within mineral structures is referred to as isomorphous substitution and will be discussed in more detail later in this chapter.
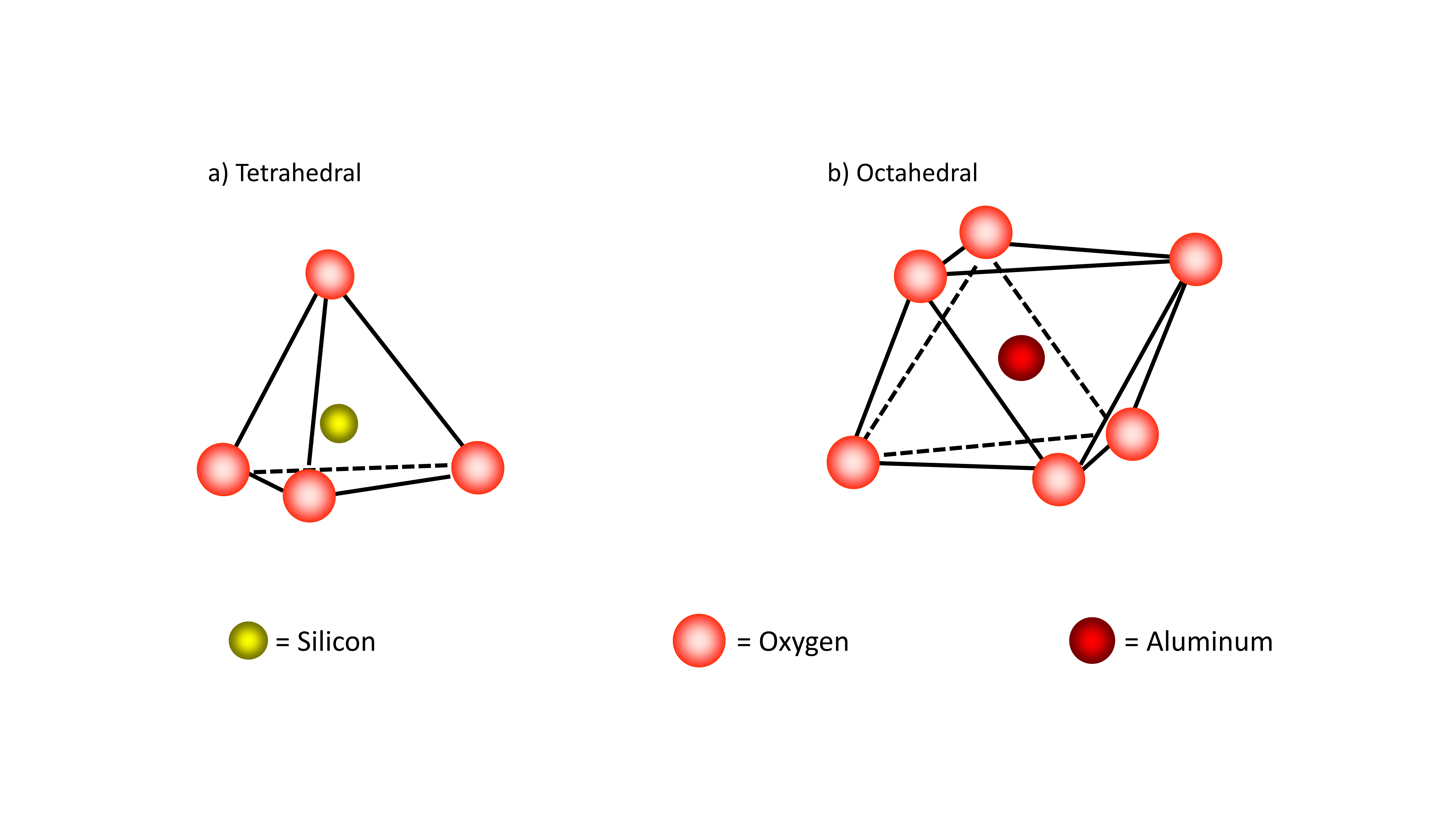
The geometry of ions about a central ion can be visualized as a coordinating polyhedron. In mineralogy, these polyhedra are usually constructed from the arrangement of anions; around the cations but sometimes the cations may be absent altogether. By far the most common polyhedral structures that make up most soil minerals are the tetrahedron (four-sided structure with four oxygen atoms coordinating around Si) and the octahedron (eight-sided structure with six oxygen atoms coordinating around aluminum; Figure 14.5). Since Si and O are by far the most abundant elements in soil, minerals with tetrahedral structures form the basis for many of the common primary minerals. Sheet-like phyllosilicates composed of combination of silica tetrahedrons and aluminum octahedrons are secondary minerals dominating the clay-size (<0.002 mm) size fraction. Phyllosilicates will be discussed in further detail later in this chapter.
Table 14.3. Radius ratios coordination numbers and associated structural units for most cations relative to structural oxygen (O2-) in minerals
| Rcation/R(O2-) Range | Coordination Number (CN) | Polyhedron Unit |
|---|---|---|
| 0.155-0.225 | 3 | Triangular |
| 0.225-0.414 | 4 | Tetrahedral |
| 0.414-0.732 | 6 | Octahedral |
| 0.732-1 | 8 | Cubic |
| >1 | 12 | Cuboctahedron |
The fundamental difference among mineral structures is the number of shared oxygen atoms that occurs between adjacent tetrahedra. Although there are several types of mineral structures and classifications; generally speaking, most common soil minerals are composed of repeating silicate tetrahedron (SiO4) units that fall into one of four simplified general types of structures:
- isolated silica tetrahedra (neosilicates);
- chains of silica tetrahedra (inosilicates);
- framework tetrahedra (tectosilicates); and,
- sheets of tetrahedra (combined with octahedral units; phyllosilicates).
Some common soil minerals listed according to the amount of shared oxygen atoms between adjacent tetrahedra are given in Table 14.4.
Table 14.4. Some of the common soil minerals
| Oxygen Sharing in Tetrahedra | Common Mineral Groups | Example Mineral Name | Generalized Ideal Composition |
|---|---|---|---|
| Neosilicates | Olivines | Fosterite | Mg2SiO4 |
| No sharing | |||
| Isolated tetrahedra | Fayalite | Fe(II)2SiO4 | |
| Inosilicates | Pyroxenes | Enstatite | MgSiO3 |
| One oxygen shared | Diopside | (Ca,Mg)SiO3 | |
| Augite | (Ca,Mg,Fe(II),Al)SiO3 | ||
| Inosilicates | Amphiboles | Tremolite | Ca2Mg5[Si8O22(OH)2] |
| Two oxygen shared | Actinolite | Ca2(Mg,Fe(II))5[Si8O22(OH)2] | |
| Hornblende | (Ca,Na)(Mg,Fe(II),Al)5[(Al,Si)8O22(OH)2] | ||
| Phyllosilicates | Kaolinite | Si2Al2O5(OH)4 | |
| Three oxygens shared | Muscovite | KAl3(AlSi3)O10(OH)2 | |
| Tectosilicates all four oxygens shared | Framework Silicates | Quartz | α-SiO2 |
| Feldspars | (Ca,Na,K)(AlxSi(3-x))O8 |
MINERALS OF THE SAND-SIZE (2 mm – 0.05 mm) FRACTION
Quartz
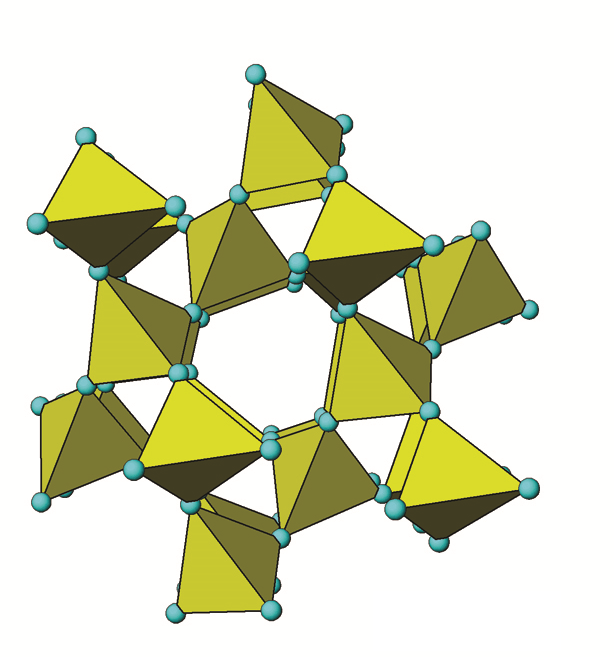
Quartz (SiO2) or more specifically, α-quartz (α-SiO2), is the most common mineral found in Canadian soils, typically accounting for the majority of the sand fraction (Figure 14.3). Quartz is a framework silicate (tectosilicate) with all four oxygen atoms in its silica tetrahedrons shared among adjacent Al,Si3 tetrahedra (Figure 14.6). It belongs to the hexagonal crystal system.
When tetrahedra share all oxygen atoms with adjacent tetrahedra, they form a very strong 3-dimensional framework of Si-O bonds. Quartz is essentially pure SiO2. The negative charge of the O atoms is exactly balanced by Si requiring no other bonding ions. Under a microscope most specimens are usually irregular shaped lustrous fragments appearing either white, milky and sometimes clear. The 3D framework chemical structure of quartz is typical of tectosilicates. The repeating units are SiO2 in a tetrahedral configuration. The Si-O bonds are very strong covalent bonds, highly resistant to weathering but overall not chemically stable in soil. The solubility of quartz is very low and the rate of weathering (dissolution) is extremely slow. Quartz is, therefore, highly persistent in soil with a slow rate of dissolution compared to most other minerals leading to a far longer persistence, with relative quantities building up over time (e.g., Ae horizons of Podzolic and Luvisolic soils). Visually quartz appears usually as clear, white, or gray glassy fragments but sometimes exists as yellow, brown, purple, pink or red fragments that can easily be seen with the naked eye or a hand lens. Quartz usually constitutes about 40-60% of the sand (2.0–0.05 mm) fraction, a major fraction of the silt-size range (0.05-0.002 mm) and is present in small amounts in the clay-size (<0.002 mm) fraction of Canadian soils as well (see Figure 14.3).
Feldspars
The feldspars are actually a group of minerals making up the second most common tectosilicate group found in soil (after quartz), typically accounting for 15-35% of the sand fraction (Figure 14.3). Feldspars make up a large portion of granitic and metamorphic rocks. Therefore, feldspars constitute a significant portion of our soil. There are many mineral species making up the feldspar group, with all having a general formula of X(Al,Si)4O8 where X is most commonly K, Na or Ca with Al and Si ions making up the tetrahedral units. Other ions such as Ba, Zn, Rb, Sr, and Fe2+ also substitute for X in feldspar structures (King 2020). Feldspars typically appear off-white, with various shades of red, orange, brown and occasionally green existing. The 3D framework of the feldspars is made up of units ranging from AlSi3O8 to Al2Si2O8 in a “kinked ribbon” configuration (Figure 14.7a). The K, Na or Ca found in the voids of the kinks with sufficient Al substituting for Si within the kinked ribbon structure to produce a negative structural charge to neutralize the positive charge on the cations.
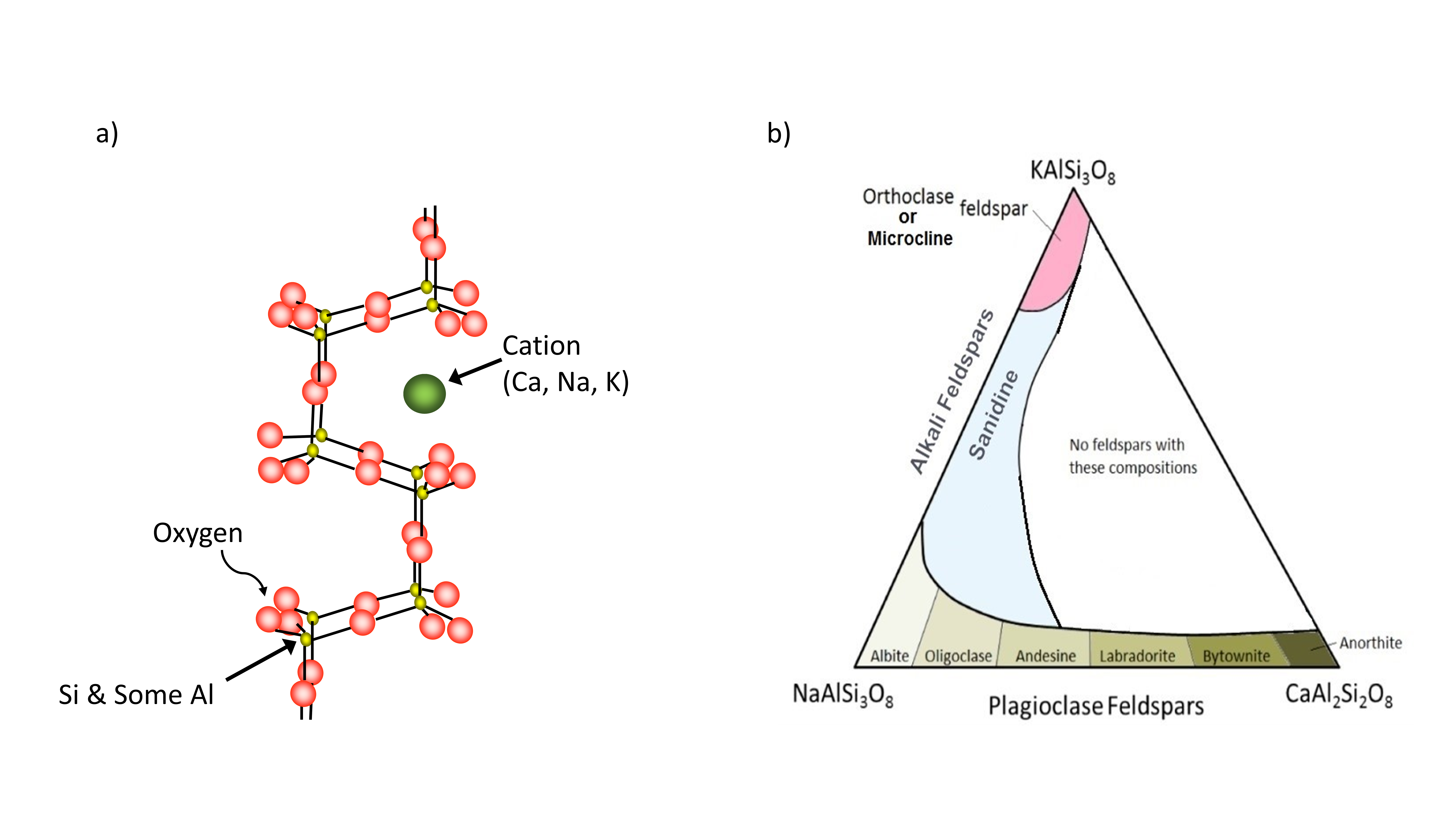
The most common feldspar minerals and their ranges in composition are illustrated in the ternary diagram (Figure 14.7b). Orthoclase and microcline are the mineral names given to feldspars with the same chemical composition dominated by potassium (KAlSi3O8) with minor amounts (up to about 25%) of Na substituting for K in the structure (Figure 14.7B). Orthoclase and microcline are polymorphs, which means they have the same chemical composition but different crystal forms. Orthoclase has a monoclinic crystal form while microcline is triclinic. Feldspars containing more than about 25% Na but less than 75% K constitute the mineral sanidine [(K,Na)AlSi3O8]. Feldspars within the full range of K and Na compositions (orthoclase/microcline, sanidine to albite) are generally referred to as the alkali feldspars (Figure 14.7B). Sodium and Ca feldspars occur in a continuous series called the plagioclase series with compositions ranging from NaAlSi3O8 to CaAl2Si2O8 where the charge on the amount of Na and Ca in the structure is neutralized by substitution of Al for Si in the tetrahedral kinks. Plagioclase feldspars with specific compositions are given specific mineral names listed in Table 14.5. The plagioclase series of minerals is an example of a solid solution series where composition varies between two end-member minerals (i.e., albite; NaAlSi3O8 and anorthite; CaAl2Si2O8) that share the same basic chemical formula but experience substitution of elements in one or more atomic sites.
Quartz and the feldspars have similar specific gravity and hardness, thus they tend to be transported and deposited together. Unlike quartz, feldspars have several cleavage planes along which the minerals fracture— making them brittle and more prone to mechanical breakdown upon impact during physical transport. These cleavage planes are weakness planes along which water and microbial exudates may infiltrate and accelerate their chemical alteration. Organisms such as bacteria and fungal hyphae also infiltrate these weakness planes to enhance feldspar degradation. Hence, feldspars tend to weather and dissolve at a faster rate compared to quartz (Cousineau, 2020).
Table 14.5. Range in composition for plagioclase feldspars
| Mineral Name | % NaAlSi3O8 | % CaAl2Si2O8 | Range of Composition |
|---|---|---|---|
| Albite | 90-100 | 0-10 | Na(Al,Si3O8) – Na0.9Ca0.1(Al1.1Si2.9O8) |
| Oligoclase | 70-90 | 10-30 | Na0.9Ca0.1(Al1.1Si2.9O8) – Na0.7Ca0.3(Al1.3Si2.7O8) |
| Andesine | 50-70 | 30-50 | Na0.7Ca0.3(Al1.3Si2.7O8) - Na0.5Ca0.5(Al1.5Si2.5O8) |
| Labradorite | 30-50 | 50-70 | Na0.5Ca0.5(Al1.5Si2.5O8) - Na0.3Ca0.7(Al1.7Si2.3O8) |
| Bytownite | 10-30 | 70-90 | Na0.3Ca0.7(Al1.7Si2.3O8) - Na0.1Ca0.9(Al1.9Si2.9O8) |
| Anorthite | 0-10 | 90-100 | Na0.1Ca0.9(Al1.9Si2.9O8) - Ca(Al2Si2O8) |
“Heavy” Minerals
“Heavy” minerals represent a generalized group of “other” primary minerals usually found only in minor amounts (2-5%) in the sand fraction of Canadian soils. Several of these minerals are dark coloured due their high content of Fe and Mg. Others such as garnet and hematite are dark red and zircon is yellow to colorless. Heavy minerals may be isolated from the sand and silt-size fractions based on density separation (Andò, 2020). Heavy minerals are not typically found in the clay-size fraction. Heavy minerals for the most part are generally defined as having particle density (specific gravity) greater than 2.90 g/cm3 (Andò, 2020). Some common minerals found in the sand fraction of soils are provided in Table 14.6. Although the heavy mineral component is only a minor mineral component of the sand fraction, it may supply many critical plant nutrients through progressive weathering.
Once isolated, the heavy mineral fraction may be examined by several methods including light and electron microscopy coupled with computer machine learning techniques (Maitre et al., 2019), powder X-ray diffraction, and other physical or chemical methods. Heavy minerals generally include mafic minerals typically with high concentrations of Fe and Mg. Macroscopically pyroxenes and amphiboles can be difficult to distinguish from one another, as they are both dark-colored. Typically, pyroxene crystals are shorter crystals while amphiboles have longer needle-like crystals. Other minerals in the heavy mineral fraction may include garnets (X3Al2Si3O12 were X may be Mg, Fe(II), Mn, or Ca), magnetite (Fe3O4), chromite (Fe,Cr(III)2O4), hematite (Fe2O3), zircon (ZrSiO4), rutile (TiO2).
Table 14.6. Some common minerals in the sand-size fraction with associated densities
| Density Fraction | Mineral | Density (g cm-3) |
|---|---|---|
| “Light” Fraction (Density <2.9 g cm-3) |
quartz | 2.65 |
| feldspars | 2.55-2.76 | |
| “Heavy” Fraction (Density >2.9 g cm-3) |
olivine | 3.22-4.39 |
| augite (pyroxene) | 2.96-3.52 | |
| hornblende (amphibole) | 3.02-3.45 | |
| hematite (Fe2O3) | 5.26 | |
| magnetite (Fe3O4) | 5.2 |
MINERALS OF THE SILT-SIZE (0.05 – 0.002 mm) FRACTION
The silt-size fraction of soils is intermediate in size and mineralogy between the sand and clay-size fractions. Hence, the silt fraction effectively contains a physical mixture of minerals found in both the sand-size fraction and the clay-size fraction. Readers are therefore directed to those sections of this chapter for further discussion.
Can You Dig It!
The terms “clay” and “clay mineral” are often used interchangeably and see commonplace use in the scientific literature; however, their use can be ambiguous and sometimes misleading. The term “clay” is typically used to refer to a clay soil or the clay-size fraction (particles <0.002 mm diameter). “Clay minerals” refer specifically to phyllosilicates which account for the majority of “clay” in soil but does not include oxyhydroxides and amorphous aluminosilicates. Although not phyllosilicates, numerous sand-size mineral described above may also be present in the clay-size fraction.
MINERALS OF THE CLAY-SIZE (<0.002 mm) FRACTION
As discussed previously, the clay-size fraction is comprised mostly of secondary hydrated aluminosilicates. These minerals are very fine-grained and thus have a very high SSA and account for the largest portion of the available and reactive mineral surface area in soil (Chapter 5, Table 5.1). In addition, minerals in the clay fraction have both permanent and pH variable charge capable of attracting and retaining plant nutrients with direct and indirect impacts on the soil fertility. In short, they are the most chemically active inorganic phases in soil.
SHEET SILICATES: PHYLLOSILICATES
By far the most common type of minerals in the clay-size fraction are the sheet or layer silicates called phyllosilicates (Greek; phyllon meaning leaf or sheet), which appear as flat sheet structures in photomicrographs (e.g., Beutelspacher and Van Der Marel, 1968). Phyllosilicates are crystalline minerals, with a structure consisting of layers of repeating atomic units; thus given the name “layer” or “leaf” aluminosilicates. The fundamental layers are composed of tetrahedral and octahedral sheets that occur in different combinations to form different phyllosilicate mineral species.
Tetrahedral sheets are composed of repeating tetrahedron (four-sided) units each containing a Si atom surrounded by four O atoms. Three of the four O atoms are shared with adjacent tetrahedral units, referred to as the basal O atoms. The one unshared O is referred to as the apical O (Figure 14.8). The shared basal O atoms combine to form sheets that continue in two dimensions ad infinitum.
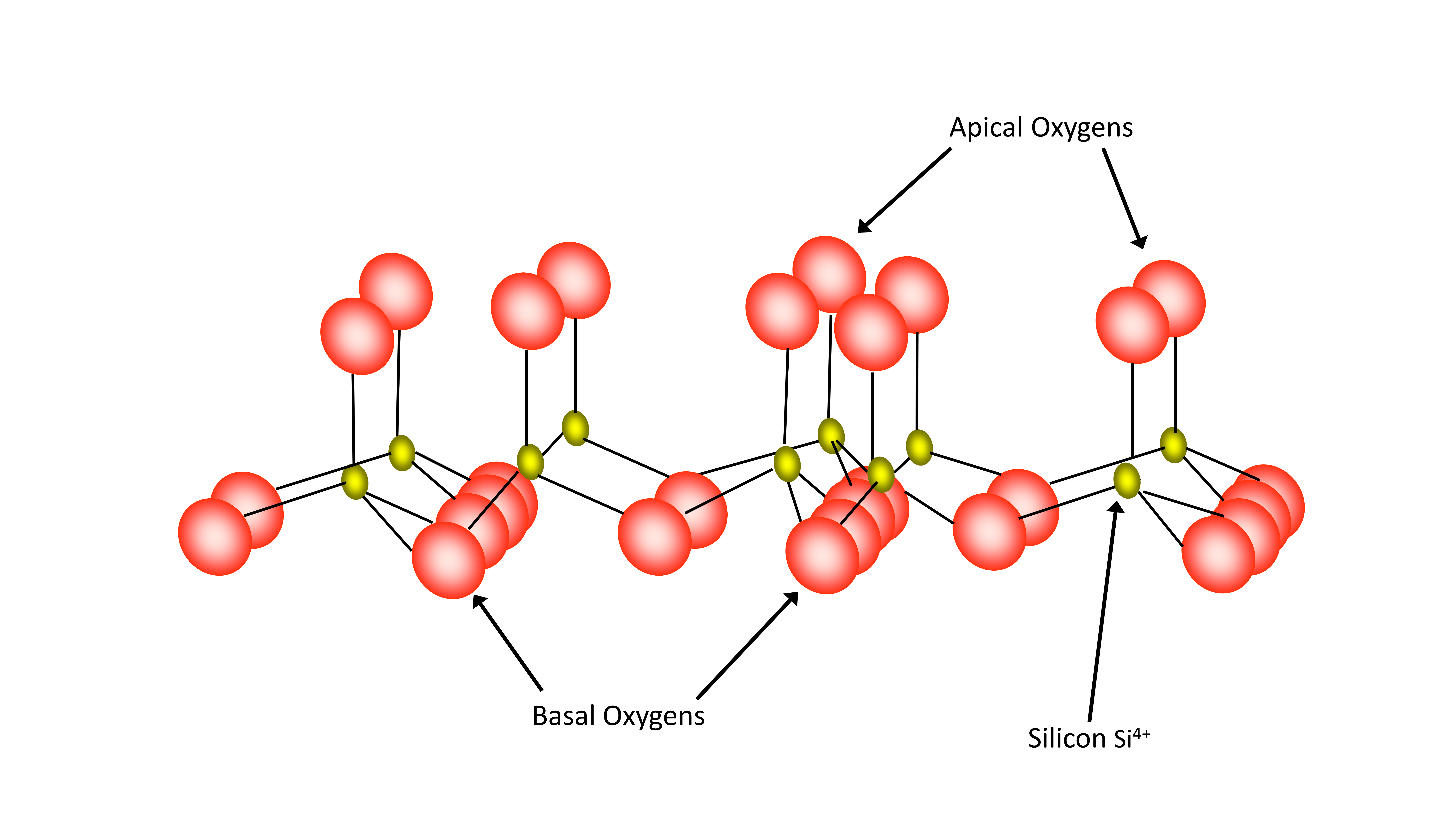
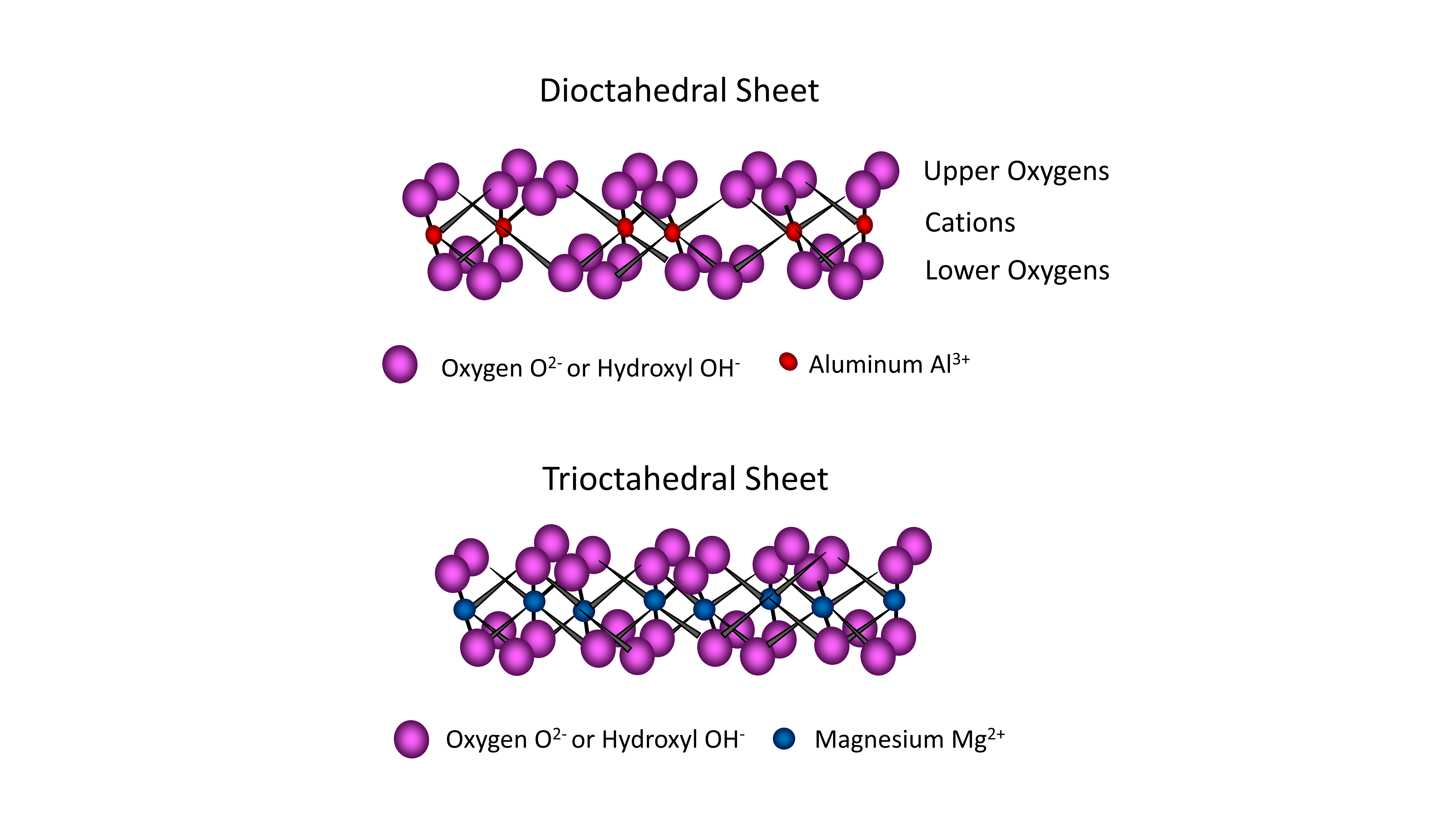
The octahedral sheets found in phyllosilicates are composed of multiple octahedron units, each unit containing Al and/or Mg atoms surrounded by six O atoms or OH– groups. Three O atoms (or OH– groups) occur in the lower plane, with three oxygen atoms (or hydroxyl groups) in the upper plane sandwiching aluminum or magnesium atoms between, forming an eight-sided (octahedral) configuration and a pseudohexagonal form. These units combine by sharing O atoms to form an octahedral sheet extending in two dimensions ad infinitum (Figure 14.9). When the central cation of the octahedron is trivalent, such as aluminum (Al3+), only two of three (2/3) cation sites in an octahedral sheet require filling in order to neutralize the charge by the surrounding O atoms or OH– ions. In this case, the octahedral sheet is referred to as dioctahedral (Figure 14.9). When the central cation of the octahedron is divalent, such as Mg2+, then all three of three (3/3) cation sites in an octahedral sheet require filling in order to neutralize the negative charge of the surrounding O or OH–. In this case, the octahedral sheet is referred to as trioctahedral (Figure 14.9).
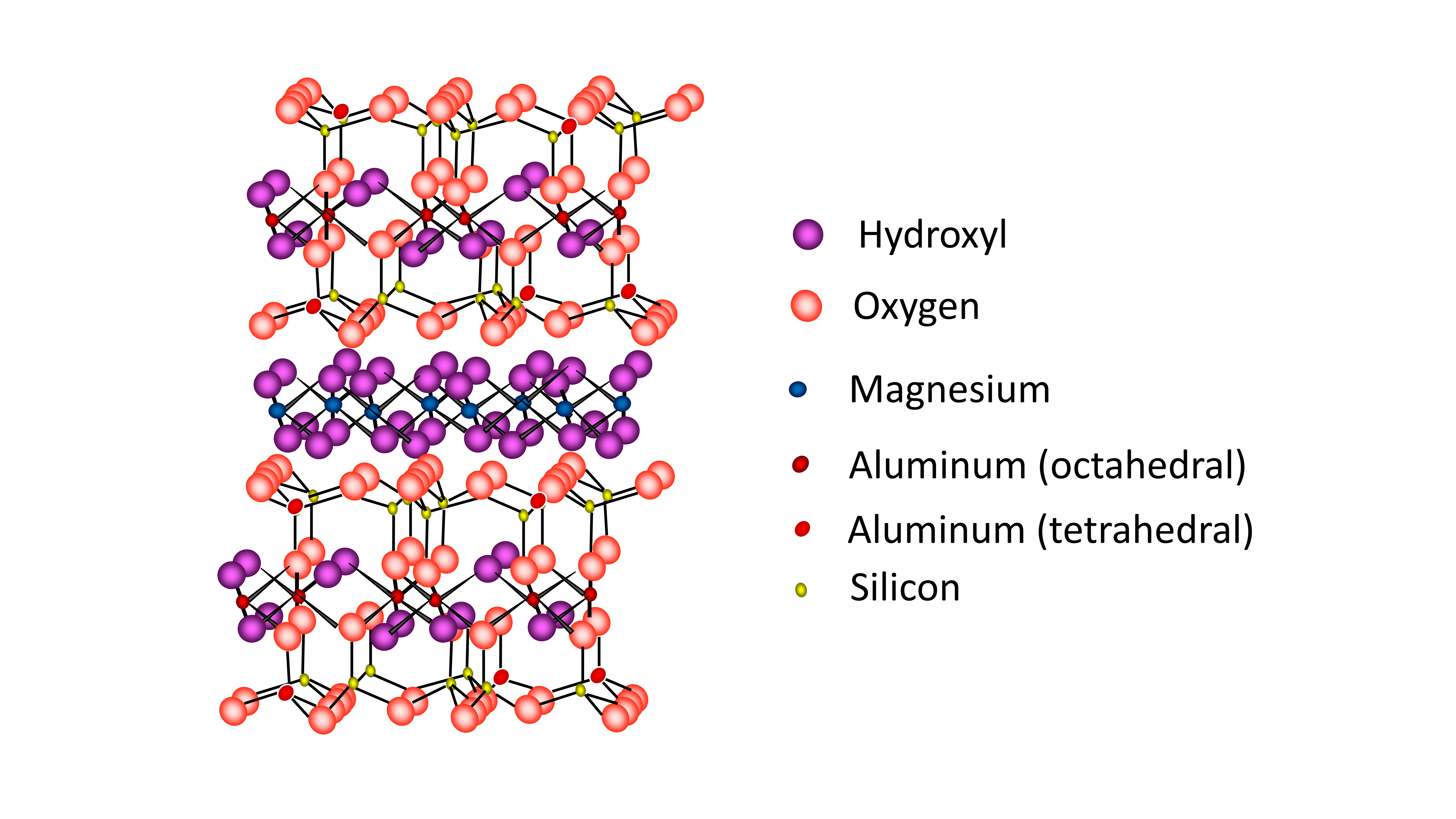
Phyllosilicate structures have tetrahedral and octahedral sheets combined to form layers through sharing of the apical O atom of the tetrahedral sheet with O atoms in the octahedral sheet. Phyllosilicates composed of one tetrahedral sheet and one octahedral sheet are called 1:1 type phyllosilicates. Those composed of two tetrahedral sheets sandwiching one octahedral sheet between are called 2:1 type phyllosilicates. Phyllosilicates consist of many layers (1:1 type or 2:1 type) stacked one on top of the other with some form of bonding between the layers. The region located between two adjacent 1:1 or 2:1 layers is referred to as the interlayer region. Some 2:1 type phyllosilicates may also contain an additional octahedral sheet composed completely of hydroxides in the interlayer region. These are referred to as 2:1:1 or 2:2 type phyllosilicates. The basic behaviour and properties of the different phyllosilicates are dictated in large part by the characteristics associated with the interlayer region of the clays found in a given soil.
Isomorphous Substitution
In addition to their extremely large SSA, phyllosilicates are also electrically charged due to the substitution of the central cations within the tetrahedral or octahedral sheets for others of similar size but different valence (Table 14.2). Table 14.3 provides ranges of radius ratios corresponding to common coordination numbers and their corresponding structural units. Ions are typically interchangeable within a structure if their size does not differ by more than about 15% and valences do not differ by more than one unit. Substitution occurs at the time the phyllosilicates form. The substitution of a cation of lower valence for another of similar size imparts a net negative charge to the structure of the mineral because the charge on the surrounding O atoms is not balanced completely. For example, trivalent aluminum (Al3+) may substitute for some of the tetravalent silicon (Si4+) atoms in the tetrahedral sheet. The lesser charge of the Al compared to Si creates a charge deficit with a resulting net negative charge within the tetrahedral sheet of the structure. Similarly, in dioctahedral sheets, Mg2+, Fe2+, or other divalent cations, may substitute for some Al3+ resulting in an extra negative charge associated with the octahedral sheet of the structure. Similarly, Li+, although occurring in low abundance (Table 14.1), also sometimes substitutes for Mg2+ in trioctahedral phyllosilicates (lepidolite).
To neutralize the negative charges produced by isomorphous substitution within the structure of phyllosilicates, positively charged cations are attracted from the soil solution to the surface of the minerals. These charged neutralizing cations that migrate into the interlayer region of the phyllosilicates are called exchangeable cations. These cations are capable of exchanging with other cations in the soil solution. Collectively, the overall abundance of negative charge on the clay structure is referred to as the cation exchange capacity (CEC). The abundance of negative charge sites (the charge density); the location of the negative charges (tetrahedral and/or octahedral sheet), and the type of cation in the interlayer region neutralizing the charge (e.g., Na+, Ca2+, Mg2+, K+ or other) all determine the ability of the phyllosilicate to be expandable (swelling clay), and control the capacity to retain soil and plant nutrients.
The Interlayer Region
In addition to attracting and retaining cations, the nature of the interlayer region of some phyllosilicates may also be controlled by hydrogen bonding. In 1:1 type clay minerals, hydroxyl groups associated with the octahedral sheet in the structure face a layer of O atoms formed by the apical Os in the adjacent 1:1 layer (Figure 14.10). The result is that hydrogen bonding holds the 1:1 layers closely together forming a very stable structure. Consequently, all 1:1 type phyllosilicates do not expand and are thus non-swelling.
In contrast, hydrogen bonding does not occur in 2:1 clay minerals as basal O atoms in two adjacent layers face each other with no OH– groups (Figure 14.11). Thus, the unit layers are held together by the weaker electrostatic forces associated with whatever neutralizing cation occupies the interlayer region to neutralize the negative charges associated with isomorphous substitution within the layer structures. The interlayer cations may be any combination of Ca2+, Mg2+, Na+, K+, NH4+, plant nutrients (Zn2+ Cu2+, Mn2+, Ni2+), iron (Fe2+, Fe3+), aluminum (Al3+) or other cations. These cations are typically exchangeable and have some amount of water associated with the cation.
The degree of expandability of a 2:1 clay mineral depends on the total layer charge; the location of the layer charge (tetrahedral vs. octahedral sites) and the nature of the exchangeable cation. The 2:1 phyllosilicates with a low layer charge (<0.6 per formula unit)—usually associated with the octahedral sheet—typically exhibit the capability of swelling. Swelling properties are usually associated with the smectites (see Table 14.7). The 2:1 phyllosilicates with a higher layer charge (>0.6 per formula unit)—usually associated with the tetrahedral sheets—are far less likely to exhibit swelling properties. The non-swelling 2:1 phyllosilicatess are the vermiculites and micas (Table 14.7).
Water is a polar molecule, allowing development of hydration spheres with the exchangeable cation. Divalent cations such as Ca2+ and Mg2+ typically have two layers of water associated with it in the interlayer region of 2:1 clay minerals with lower total layer charge. Large monovalent cations such as Na+ and K+ obviously, having a lower charge, are not as capable as divalent cations at holding the 2:1 structures together and therefore may allow more water molecules to be incorporated into the interlayer region. Under these conditions the 2:1 structures separate to many times their original (“dry”) configuration and the clay is said to be a swelling clay. This is typical of the smectite group of 2:1 phyllosilicates (see below). Note that in addition to water, polar organic molecules can also migrate into the interlayer region.
In some instances, the interlayer cation in 2:1 type phyllosilicates may become hydroxylated in six-fold (octahedral coordination). Such is the case with Al, Mg and Fe. In these cases, the interlayer becomes filled with an additional octahedral sheet which does not share O atoms with the main 2:1 layer but is hydrogen-bonded to the adjacent oxide surfaces forming a phyllosilicate which has a 2:1:1 (or 2:2) type structure. These types of phyllosilicates are referred to as the chlorite group or, more generally, chlorite.
Common Phyllosilicates
There are about 50 different species of phyllosilicates but most are rare or occur in soils found in association with specific geological deposits (e.g., halloysite in volcanic ash soils in humid regions). A general classification scheme for some of the more common phyllosilicates is shown in Table 14.7. The five species of phyllosilicates (kaolinite, smectite (montmorillonite and beidellite), micas (including hydrous mica or illite), vermiculite and chlorite) commonly identified in Canadian soils are discussed in detail below.
Table 14.7. A general classification scheme for some phyllosilicates
| Type | Group x = charge per formula unit |
Subgroup | Example Species | Idealized formula unit* |
|---|---|---|---|---|
| 1:1 | Kaolinite x~0 |
Kaolinite | Kaolinite | Si2,Al2O5(OH)4 |
| Halloysite | Si2,Al2O5(OH)4 2H2O | |||
| Serpentines | Serpentine | Si2,Mg3O5(OH)4 | ||
| 2:1 | Pyrophyllite-talc x~0 |
Pyrophyllite | Pyrophyllite | Si4,Al2O10(OH)2 |
| Talc | Talc | Si4,Mg3O10(OH)2 | ||
| Smectite x ≈ 0.25–0.6 |
Dioctahedral | Montmorillonite | 0.33M+Si4(Al1.67,Mg0.33)O10(OH)2 | |
| Beidellite | 0.33M+(Si3.67Al0.33)(Al2.00)O10(OH)2 | |||
| Nontronite | 0.33M+(Si3.67Al0.33)(Fe(III)2.00)O10(OH)2 | |||
| Trioctahedral | Saponite | 0.33M+(Si3.67Al0.33)(Mg3.00)O10(OH)2 | ||
| Hectorite | ||||
| Vermiculite x ≈ 0.6–0.9 |
Dioctahedral | Dioctahedral vermiculite | 0.86M+(Si3.47Al0.53)(Al1.67Mg0.33)O10(OH)2 | |
| Trioctahedral | Trioctahedral vermiculite | 0.86M+(Si3.47Al0.53)(Al1.67Mg0.33)O10(OH)2 | ||
| Micas x ≈ 1 |
Dioctahedral | Muscovite | K1.00(Si3.00Al1.00)(Al2.00)O10(OH)2 | |
| Paragonite | ||||
| Trioctahedral | Biotite | K1.00(Si3.00Al1.00)(Fe(II)3.00)O10(OH)2 | ||
| Phlogopite | ||||
| 2:1:1 | Chlorite x = variable |
Di-Dioctahedral | Donbassite | (Al2(OH)6)(Si,Al)4Al2O10(OH)2 |
| Tri-Dioctahedral | Sudoite | (Mg3(OH)6)(Si,Al)4 Al2O10(OH)2 | ||
| Di-Trioctahedral | Ferri-sudoite | (Al2(OH)6)(Si,Al)4(Mg,Fe)3O10(OH)2 | ||
| Tri-Trioctahedral | Clinochlore | (Mg3(OH)6)(Si,Al)4(Mg,Fe)3O10(OH)2 | ||
| *The sequence of elements within the idealized formula unit follows the order: exchangeable cation (if any) or interlayer octahedral sheet; tetrahedral cations; octahedral cations; oxygens and hydroxyls. Within each sheet the major cation (e.g. silica) is followed by the isomorphically substituting cation(s). | ||||
Kaolinite is a 1:1 type clay mineral constructed of one tetrahedral sheet and one octahedral sheet (Figure 14.10). The 1:1 layers are held together by hydrogen bonds between the layers so that structure is non-expandable as water molecules cannot enter the interlayer region. Kaolinite exhibits very little swelling when wet, or shrinkage on desiccation. The interlayer is not exposed and therefore the specific surface area is low. The mineral has almost no isomorphous substitution; therefore, these kaolinite minerals have a low surface charge and low CEC, although they have minor variable charge sites at the edges. Kaolinite is the most common 1:1 type phyllosilicate and is ubiquitous in Canadian soils.

Smectite is a freely expandable 2:1 type phyllosilicate (Figure 14.11). There are two common species of smectite: montmorillonite and beidellite. The amount of isomorphous substitution in both minerals is moderate compared to other 2:1 type phyllosilicates. The layer charge for smectites is in the range of about 0.25 to 0.6 sites per formula unit (Mx+(Si,Al)4(Al,Mg)2O10(OH)2) where Mx+ generally refers to exchangeable cations (e.g., Na+, Mg2+, K+, Ca2+, etc.). Montmorillonite and beidellite fall within the same range of total layer charge but are differentiated based on the location of the layer charge in the structure (Table 14.7). Most of the layer charge in montmorillonite is seated in the octahedral sheet while most is associated with the tetrahedral sheet of beidellite. Specific surface area is high (about 800 m2 g-1) and both water molecules and cations can enter the interlayer space.
Smectite-rich soils are sticky when wet and form large cracks under dry conditions. Heavy clay textured soils containing >60% clay of which at least half is smectite are defined as Vertisols in Canada (SCGW, 1998). These soils are described as “self mulching” as repeated wetting and drying cycles form slickenslides, which are polished surfaces on opposing ped surfaces along soil fissures formed where soil materials move back and forth past each other as large continuous cracks reopen and close with wetting and desiccation. The soil forming process causing the formation of slickenslides is called argilli-pedoturbation, being especially common in Vertisolic soils formed in the smectite-rich glacio-lacustrine parent materials found throughout the Prairie Provinces.

Micas, including hydrous mica/illite are a 2:1 type clay minerals with two tetrahedral sheets sandwiching an octahedral sheet. The 2:1 layers are held tightly together by large K+ in the interlayer region (Figure 14.12). The structure of the micas is similar to that of the smectites except that the layer charge is much higher (about 1 per formula unit) and isomorphous substitution occurs almost exclusively in the tetrahedral sheets. Potassium almost exclusively occupies the interlayer region. The very high layer charge, combined with the location of the charge in the tetrahedral sheets very close to the interlayer region, prevents expansion of the mineral. Micas are primary minerals derived from granite and metamorphic rocks of the Canadian Shield. Hydrous mica/illites form in soil from the partial weathering of the micas. Micas and hydrous mica/illite are found in almost all soils in Canada. There are two common species of mica differentiated by the nature of the octahedral sheet, namely dioctahedral muscovite and trioctahedral biotite (Table 14.7).
Hydrous mica/illite is a partial weathering product of the micas, with some (up to 20-30%) of the interlayer K+ being removed from the interlayer region and replaced with other exchangeable cations. There is also loss of tetrahedral Al3+ from the structure. Hydrous mica/illite is usually produced through the weathering of dioctahedral muscovite whereas the trioctahedral biotite structure typically breaks down totally due to relative ease of chemical attack on iron (Fe(II)) in the structure.
Although the basic structure of the micas and hydrous mica/illite are similar to smectite, the properties are very different primarily due to the higher amount of isomorphous substitution in the tetrahedral sheet, where Al3+ is substituted for Si4+ (Figure 14.12). The strong negative charge located in the tetrahedral sheets close to the interlayer surface is balanced by K+, locking the adjacent layers together. Thus, the mineral has limited expansion as water molecules cannot enter the interlayer space. Since K+ are trapped in the lattice preventing expansion, the CEC is low.

Vermicullite is a 2:1 type phyllosilicate that is similar structurally to smectite and the micas. The layer charge is in the range of 0.6 to 0.9 sites per formula unit (Mx+(Si,Al)4Al2O10(OH)2), which is intermediate between the micas and the smectites (Figure 14.13). As in the micas, the layer charge is located almost exclusively within the tetrahedral sheet. The expandability of vermiculite is limited and it has moderate swelling and shrinkage characteristics. With a combination of a layer charge lower than the micas but higher than smectite and with most isomorphous substitution is in the tetrahedral sheet vermiculite has a high surface charge and a high CEC. As the structure is similar to the micas, vermiculite is capable of trapping K+ ions in the interlayer region reverting the structure to partly resemble hydrous mica/illite.
Chlorite is the name for a mineral, as well as a group of 2:1:1 (or 2:2) type phyllosilicates constructed of a 2:1 layer structure with a hydroxide layer in the interlayer region (Kohut and Warren 2002). The mineral chlorite is the most common mineral species within the chlorite group. The structure is composed of an Al, Mg or Fe hydroxide layer in six-fold (octahedral) coordination in the interlayer region between 2:1 type layers (Figure 14.13). This interlayer hydroxyl layer does not share O atoms with the main 2:1 layer. The hydroxide layer is hydrogen bonded to the adjacent 2:1 layers and is non-expandable (Figure 14.13). The amount of isomorphous substitution is variable, but as the structure is non-expandable so the overall CEC is low.
Exchange Capacity and Specific Surface Area
The clay-size fraction of most Canadian soils typically contain a mixture of the five common phyllosilicates along with small amounts of amorphous aluminosilicates and Fe and/or Al oxy-hydroxide. Proportions are mostly a function of the origins of the soil parent materials. Variations in the overall ion exchange and adsorptive properties of soils depends on the mix of phyllosilicates present. The two main differences among the phyllosilicates is their CEC and the amount of reactive surface area are summarized in Table 14.8. Kaolinite has very little isomorphous substitution and is non-expanding. Consequently, it has a very low CEC and all surface area is associated with the external surfaces of the structure. Hydrous mica/illite and the chlorites have a significant amount of isomorphous substitution; however as their interlayer regions contain either tightly bound K+ or an additional hydroxide layer, respectively, which inhibits layer expansion. Thus, CEC is limited and the structures are non-expandable. Smectites have an intermediate amount of isomorphous substitution coupled with the ability to admit exchangeable cations into the interlayer region. Consequently, both CEC and total surface area is high. Soils containing smectites are noted for their high plasticity, cohesion and marked ability to swell when wet and shrink when dried. Vermiculites have more isomorphous substitution compared to smectites and thus have a higher CEC; however, most of the charge in vermiculites is concentrated within the tetrahedral sheets closer to the interlayer region and therefore has a greater electrostatic attraction for exchangeable cations in the interlayer region. As a result, although the total surface area of vermiculite is similar to the smectites, layer expansion of vermiculite structures is limited due to restricted entry of water into the interlayer region.
Table 14.8. Ranges of cation exchange capacity (CEC) and internal and external surface area in common phyllosilicates and other colloids. (Adapted from Weil and Brady, 2017)
| Phyllosilicate/Colloid | CEC (cmol(+) kg-1) |
External Surface Area (m2 g-1) |
Internal Surface Area (m2 g-1) |
|---|---|---|---|
| Kaolinite | 2-16 | 5-15 | ~0 |
| Chlorite | 10 - 40 | 5-20 | ~0 |
| Hydrous Mica/Illite | 20 - 40 | 100 - 125 | ~0 |
| Smectite | 60 - 120 | 80 - 140 | 570 - 660 |
| Vermiculite | 100 - 150 | 70 - 120 | 600 - 700 |
| Amorphous aluminosilicates | 5 - 350 | 500 - 1450 | ~0 |
| Fe/Al | ~0 - 3 | 200 - 500 | ~0 |
| oxy-hydroxides |
Can You Dig It!
Although not confirmed, some data suggest that phyllosilicates could be a template to synthesize RNA from simple components and was the catalyst for the abiotic origin of life itself on this planet (Berman, 2019). Clay minerals act as very efficient catalysts in the polymerization of amino acids and nucleotides. RNA adsorbed to clay minerals can be encapsulated within vesicles. Once formed, such vesicles could grow and divide by incorporating fatty acids thus mediating vesicle replication through cycles of growth and division (Brack, 2013)
DISTRIBUTION OF PHYLLOSILICATES IN CANADIAN SOILS
The distribution of phyllosilicates in Canadian soil varies somewhat by physiographic region (Kodama, 1979); most phyllosilicates are inherited from the parent materials deposited by the retreat of the continental glaciers during the late Pleistocene. For example, some clay minerals such as kaolinite, mica, (i.e., biotite and muscovite) and chlorite are inherited directly from the Canadian Shield. Others in the Cordilleran region and Interior Plains Regions are derived from glacial degradation of sediments from the Cretaceous period (65 million years ago). Comparison of data for clay-size separates of soils, for example, from the Interior Plains of western Canada with those found in the eastern Canada (St. Lawrence Lowlands) show a distinct difference between the suite of clay minerals (Table 14.9). The Interior Plains region of western Canada is commonly reported to contain smectite, clay-size mica, chlorite and some kaolinite, (Kodama, 1979; Dudas and Pawluk, 1982; Spiers et al. 1989a; Spiers et al. 1989b; Warren and Dudas, 1992). In contrast, the clay-size fraction of soils from the Western St. Lawrence Lowlands (Ontario) derived from much older sediments formed during the Devonian (359 million) and Silurian (443 million) periods and are dominated by clay-size mica, chlorite, vermiculite and kaolinite with some reports of traces of smectite (Kodama, 1979). Parent materials in both regions had contributions from the Canadian Shield. There is only weak evidence of alteration in the diverse clay mineral assemblages from the various physiographic regions and correlation is futile between generic types of soil. An exception is the Podzolic soils and to a lesser extent Brunisolic and Gleysolic soils where inherited chlorite minerals appear to weather to expandable phyllosilicates (Kodama, 1979).
Table 14.9. Range of abundances for phyllosilicates typically found in the clay-size fractions of soils of the Interior Plains and from the Western St. Lawrence Lowlands
| Interior Plains Region | St. Lawrence Lowlands | |||
|---|---|---|---|---|
| Phyllosilicate | g kg-1 | Phyllosilicate | g kg-1 | |
| Kaolinite | 70-110 | Kaolinite | 120-150 | |
| Discrete Muscovite | 250-340 | Discrete Muscovite | 350-450 | |
| Montmorillonite | 470-490 | Vermiculite | 180-220 | |
| Chlorite | 120-160 | Chlorite | 240-300 | |
| Vermiculite | trace | Smectite | trace | |
Amorphous Aluminosilicates
Other hydrous aluminosilicates are also found in soil but are different structurally from the phyllosilicates. These are described as amorphous (lacking form or shape) or nanocrystalline and do not have a definite ordered crystalline structure like the phyllosilicates. Allophane and imogolite are examples of amorphous aluminosilicate minerals, commonly formed during rapid weathering in volcanic ash soils found in humid climates of countries such as New Zealand, Chile, and Japan. Volcanic ash weathers rapidly and typically dissolves releasing large amounts of soluble aluminum and silica, which can precipitate as allophane or imogolite. Amorphous aluminosilicates are non-expandable, but are extremely small (<4 nm diameter) with an extremely high specific surface area (700-1500 m2 g-1) and very high ion exchange capacities. Although they occur in abundance in volcanic ash soils, they may occur in small amounts in some soils along with phyllosilicates. Imogolite, for example, is documented in some Humo-ferric Podzolic soils in British Columbia (Grand and Lavkulich, 2013; 2015); elsewhere in Canada (McKeague and Kodama, 1981; Wang et al., 1991) and Solodized Solonetzic soils in Alberta (Spiers et al., 1984).
IRON AND ALUMINUM OXYHYDROXIDES
Oxyhydroxide clays (sesquioxides) are secondary oxides, hydroxides and hydrous oxide minerals of Fe, Al, and Mn. Some are well crystallized (e.g., gibbsite, hematite, goethite), while others are amorphous. These minerals are the usual source of red, yellow, brown, and blueish hues observed in soil. Crystalline oxide clays are composed of sheets of oxygen and/or hydroxyl groups in an octahedral arrangement with Fe, Al or Mn. They do not have tetrahedral sheets of Si in their structures. The octahedral structures of these minerals are held together by hydrogen bonding, therefore they are non-expanding. Iron oxides and hydroxides once called “limonite” were later found to actually be a mixture of Fe oxyhydroxide minerals.
These minerals do not carry identifiable permanent negative charges, but do have a high surface charge associated with surface hydroxyl groups. As such, the exchange capacity of oxyhydroxides is highly pH dependent with the variation in surface charges due to protonation of hydroxyl groups at low pH producing positive charges and dissociation of hydroxyl groups at high pH producing negative charges. Oxyhydroxide minerals are referred to as being amphoteric due to their variable CEC. These minerals typically have a net CEC at high pH and a net anion exchange capacity at low pH ranges.
Oxyhydroxides are usually found in small quantities in Canadian soils (Grand and Lavkulich, 2013; 2015; Spiers et al., 1984). They are highly reactive with a very high specific surface area. Presence of these minerals significantly influences soil properties such as ion adsorption and exchange. For example, Fe oxyhydroxides are capable of adsorbing and retaining large amounts of phosphate, molybdate and borate ions from the soil solution as well as scavenging metals such as Cu, Pb, Zn, Co, Cr, and Ni. Some common Fe oxyhydroxides are listed in Table 14.10.
Table 14.10. Some common secondary iron oxyhydroxides found in soil
| Mineral | Formula | Notes |
|---|---|---|
| Goethite | α-FeOOH | Most common and abundant iron oxyhydroxides in soils. Yellow to brownish yellow. Very fine particle size. Precipitates quickly from Fe3+ in solution. Very small (<0.5 um) needle-like crystals. |
| Lepidocrocite | γ-FeOOH | Found in poorly drained soils as bright orange mottles but NOT common. Formed through oxidation of precipitated Fe(II)(OH)2 in the absence of CO2 at pH 5-7. Extremely small (0.1-0.5 um) lath shaped crystals. |
| Hematite | α-Fe2O3 | Second in abundance to goethite. Prevalent in tropical soils. Forms through dehydration of goethite and ferrihydrite. Very small amounts impart a red colour to soil. |
| Ferrihydrite (aka hydrous ferric hydroxide) |
(Fe(III))2O3·0.5H2O but also 5Fe2O3·9H2O |
Highly variable composition. Precipitates directly from Fe rich oxygenated water or bacteria through biological activity. Reddy-orange. Forms nanocrystals. Metastable. |
CARBONATES
Some common carbonate minerals found in soil are presented in Table 14.11. The majority of soil carbonates originate from primary sources, being inherited from sedimentary rocks, and some are secondary precipitates usually found in subsurface horizons. The crystal structure of many carbonate minerals reflects the trigonal symmetry of the carbonate ion which usually occurs in combination with Ca, Na, U, Fe, Al, Mn, Ba, Zn, Cu, Pb, and other elements. There are about 80 known carbonate minerals; however, by far most common carbonate minerals found in Canadian soils are calcite (CaCO3) and dolomite ((Ca,Mg)CO3) derived primarily from Paleozoic bedrock. Most carbonate minerals typically contain trace amounts of other carbonate related elements (see above) substituted into their structures.
Calcite is sparingly soluble, but solubility increases at lower soil pH values and is not usually found in soils with pH values less than 6.0. Calcite typically dissolves from the solum and is leached to become re-deposited in the lower soil profile, sometimes observed as secondary calcite nodules (Miller et al., 1987; Wang and Anderson, 2000). Aragonite and other calcium carbonate minerals are neither common nor stable in soils.
Table 14.11. Some common carbonate minerals
| Mineral | Ideal Formula | Notes |
|---|---|---|
| Calcite* | CaCO3 | Main mineral component in limestone. Typically unstable and dissolves in the upper solumn but can precipitate in the C horizon usually under subhumid or dryer climatic regimes. |
| Aragonite* | CaCO3 | Derived from fossil shells of geological origin. Unstable and dissolves in soil. |
| Dolomite | (Ca,Mg)CO3 | Derived from sedimentary rocks (dolostone) of geological origin. Unstable and dissolves in soil. |
| Siderite | Fe(II)CO3 | Derived from sedimentary or metamorphic rocks. Unstable in soil but very slow to dissolve. |
| Magnesite | MgCO3 | Highly soluble in soil. |
| Trona | Na2CO3 | Highly soluble in soil. Found mostly in Solonetzic soils at high pH (~9.5) |
| *Calcite and aragonite are polymorphs with the same chemical formula but different crystalline structures. | ||
Dolomite (Ca,Mg(CO3)2) is a common calcium-magnesium carbonate mineral found in soil derived from dolostone bedrock. Like calcite, dolomite dissolves from the upper profile, contributing calcium to the precipitation of secondary calcite in the lower profile while the magnesium component is mostly leached or absorbed by plant roots. Some Mg2+ may substitute for Ca2+ within secondary carbonates but the amount is minor (<10%), primarily occurring if leaching is restricted (St. Arnaud, 1979).
Carbonates impart a number of properties to soil that include:
- maintaining high base saturation;
- maintaining high exchangeable Ca and Mg;
- maintainng pH >7;
- adsorbing/precipitating PO43- ions;
- adsorbing/precipitating metals (e.g., Al, Zn, Pb, Fe, Ni, Mn); and,
- stabilizing clays and soil structure.
PHOSPHATE MINERALS
Primary Phosphate Minerals
The average crustal abundance of P is reported as 1050 mg kg-1, while the total content of P in soils is typically in the range of 400–800 mg kg-1. Phosphate can easily combine with about 30 different elements forming over 300 minerals that contain P (Bleam, 2017). Some are primary phosphate-containing minerals derived from sedimentary and igneous rocks, but most are minerals containing phosphate that have PO4 substituted onto their structures as a minor “impurity”.
The most common “pure” primary phosphate minerals are referred to by the group name “apatite” and given the general chemical formula: Ca5(PO4)3(F,Cl,OH). Apatite usually occurs usually as very small hexagonal crystals with a variety of colours including green, bluish green, brown, white, yellow, red, gray and others depending on impurities. Names for specific mineral for the three common end members of apatite are as follows:
- fluoroapatite Ca5(PO4)3F
- chloroapatite Ca5(PO4)3Cl
- hydroxyapatite Ca5(PO4)3OH
In nature, these minerals occur as admixtures, usually dominated by one of the three components (F, Cl and OH) depending on the source. Rock phosphate or phosphorite generally contains the highest concentration of these minerals and is mined mostly for the production of phosphate fertilizers. In soils, apatite can originate from almost any igneous or metamorphic rock occurring mostly as accessory phases (Nezat et al., 2007) but rarely as a major mineral phase in rock. The mineral structure may also contain substitutions with a huge variety of ions including carbonate (CO32-), arsenate (AsO42-), V, Th, Pb, Sr, Ba, U, and rare earth elements.
Secondary Phosphate Minerals
Generally phosphate in soil reacts readily with Ca2+, Al3+, and Fe2+ and Fe3+ forming a variety of secondary precipitates (Table 14.12). This includes phosphate derived from primary mineral sources and phosphate fertilizers. Iron phosphates (vivianite and strengite) are more common in sub-oxic soils where dissolved iron is more plentiful. Aluminum phosphates (varisite) are most often associated with acidic soils where phosphate first precipitates as an amorphous intermediate that eventually transforms into varisite (Lindsay et al., 1959). Secondary calcium phosphates generally occur in calcareous soils containing limestone (mineral calcite) as a calcium source (Kar et al., 2012). Secondary brushite is commonly precipitated in calcareous soils as a result of placing high concentrations of fertilizer in bands for crop production (McLaughlin et al., 2011).
Table 14.12. Some common secondary phosphate minerals that occur in soil
| Group | Mineral Name | Ideal Formula | Occurrence |
|---|---|---|---|
| Iron | Vivianite | Fe(II)3(PO4)·2.8H2O | Flooded/wet soils |
| Phosphates | Strengite | Fe(III)(OH)2(H2PO4) or FePO2·2H2O | Flooded/wet soils |
| Aluminum Phosphates | Variscite | Al(OH)2(H2PO4) or AlPO4·2H2O | Acidic soils |
| Calcium Phosphates | Brushite | CaHPO4·2H2O | Calcareous soils |
SULPHATES AND SULFIDES
Sulphide minerals are not present in well to imperfectly drained soils but may found in some poorly drained Gleysolic or Organic soils. The sulphide mineral pyrite (FeS2) is very common primary mineral in mine wastes but can occur in shales (a sedimentary rock contributing to some soils) and as a precipitate in some tidal soils in response to highly reducing conditions. Pyrite once exposed to the atmosphere is easily oxidized releasing iron and sulphuric acid:
2FeS2 + 7O2 + 2H2O → 2Fe2+ + 4H+ + 4SO42-
As reduced iron (Fe(II)) in the sulphide structure is released, it oxidizes and precipitates as an oxyhydroxide. Oxidation of reduced S combines with water to produce a strong acid (H2SO4). Anthropogenic exposition of sulfide minerals during mining operations or other excavations is primarily responsible for acid rock drainage (ARD). Soil pH values less than 2.0 may result from the oxidation of sulphides. Sulphate derived from the oxidation of sulphides or other sources form gypsum (Table 14.13) usually in response to high pH and the presence of soluble Ca.
Table 14.13. Some sulphur containing minerals
| Type | Mineral | Ideal Formula |
|---|---|---|
| Sulphides | Pyrite | FeS2 |
| Pyrrhotite | Fe(1-x)S (x = 0 to 0.2) | |
| Sphalerite | ZnS | |
| Galena | PbS | |
| Chalcopyrite | CuFeS2 | |
| Arsenopyrite | FeAsS | |
| Pentlandite | (Fe,Ni)9S8 | |
| Cinnabar | HgS | |
| Marcasite | FeS2 | |
| Sulphates | Gypsum | CaSO4·2H2O |
| Barite | BaSO4 |
SUMMARY
In summary, the parent material of most minerals soils of Canada was deposited by the physical action of glaciers. Most minerals in these soils are derived either from the Canadian Shield or reworked and redeposited from other sources during glaciations in the millennia prior to the late Wisconsin glaciation. Primary minerals composing the majority of the sand-size fraction of Canadian soils are usually dominated by quartz and feldspars, with a minor quantity of heavy minerals. Chemical weathering of the sand size fraction contributes components to the formation of secondary minerals and to nutrients for plant uptake. Phyllosilicates dominate the clay-size fraction and constitute the most chemically active inorganic phase in soil. Differences between the phyllosilicate composition of the clay-size fraction found in regions of western and eastern Canada regions are due primarily to the source of the parent materials. Silt, intermediate in size range between the sand-size and clay-size fractions, contains a physical mixture of minerals from these fractions.
REFERENCES
Ando, S. 2020. Gravimetric separation of heavy minerals in sediments and rocks. Minerals. 10(3): 273
Berman, J.J. 2019. Evolutions clinical guidebook. Translating ancient genes into precision medicine. Academic Press. 357pp.
Beutelspacher, H., and H. Van Der Marel, 1968. Atlas of electron microscopy of clay minerals and their admixtures. A picture atlas. Elsevier. 333pp.
Bleam, W.F. 2017. Soil and environmental chemistry, 2nd Edition. Academic Press. 573pp.
Brack, A. 2013. Clay minerals and the origin of life. Chapter 10.4. Developments in Clay Science. Elsevier. 507-521pp.
Bowen, H J M, 1979. Environmental chemistry of the elements. Academic Press, London, New York.
Cousineau, P.A. 2020. Personal Communication.
Dudas, M.J. and Pawluk, S. 1982. Re-evaluation of the occurrence of interstratified and other phyllosilicates in southern Alberta soils. Canadian Journal of Soil Science. 62: 61-69.
Fanning, D., Rabenhorst, M. and R. Fitzpatrick. 2017. Historical developments in the understanding of acid sulfate soils. Geoderma 308: 191-206. http://dx.doi.org/10.1016/j.geoderma.2017.07.006
Finkl C.W. 1981. Soil mineralogy. In: Mineralogy. Encyclopedia of Earth Science. Springer, Boston, MA. https://doi.org/10.1007/0-387-30720-6_133
Grand, S and L.M. Lavkulich. 2013. Potential influence of poorly crystalline minerals on soil chemistry in Podzols of southwestern Canada. European Journal of Soil Science 64(5):651-660.
Grand, S and L.M. Lavkulich. 2015. Short-range order mineral phases control the distribution of important macronutrients in coarse-textured forest soils of coastal British Columbia, Canada. Plant and Soil. DOI 10.1007/s11104-014-2372-6
Havlin, J. L, Tisdale S.L, Nelson W.L., Beaton, J.D. 2005. Soil fertility and fertilizers: An introduction to nutrient management, 7th edition. Pearson. 528pp.
Kar, G., Peak, D. and J. Schoenau. 2012. Spatial Distribution and Chemical Speciation of Soil Phosphorus in a Band Application. Soil Science Society of America Journal 76(6):2297-2306. DOI: 10.2136/sssaj2012.0146
King, H.M., 2020. Feldspar. Geology.com. https://geology.com/minerals/feldspar.shtml
Kodama, H. 1979. Clay minerals in Canadian soils; their origin, distribution and alteration. Canadian Journal of Soil Science. 59: 37-58.
Kohut, C.K. and Warren, C.J., 2002. Chlorites. In Amonette, J.E. Bleam, W.F. Shultz, D.G. and J.B Dixon (eds.). Soil Mineralogy with Environmental Applications. Soil Science Society of America Book Series. No. 7, Madison, WI. 531-553pp.
Lindsay, W.L. Peech, M. Clark, J.S. 1959. Solubility criteria for the existence of variscite in soils. Soil Science Society of America Journal. 23(5): 357-360.
Loganathan, P. 1987. Soil Quality, Physical properties of the soil. http://www.fao.org/3/AC172E/AC172E04.htm#ch4
Liu, C. Li, X. Xu, F. Huang, P.M. 2003. Atomic force microscopy of soil inorganic colloids. Soil Science and Plant Nutrition. 49(1): 17-23.
Maitre J., Bouchard K., Bédard L.P. 2019. Mineral grains recognition using computer vision and machine learning. Computers & Geosciences 130: 84-93.
McKeague, J.A. and H. Kodama. 1981. Imogolite in cemented horizons of some British Columbia soils. Geoderma 25(3-4): 189-197.
McLaughlin, M.J., McBeath, T.M., Smernik, R., Stacey, S.P., Ajiboye, B. and G. Guppy. 2011. The chemical nature of P accumulation in agricultural soils—implications for fertiliser management and design: an Australian perspective. Plant Soil 349:69–87.
Miller, J. J., Dudas, M. J. and Longstaffe, F. J. 1987. Identification of pedogenic carbonate minerals using stable carbon and oxygen isotopes, X-ray diffraction and SEM analyses. Canadian Journal of Soil Science. 67: 953-958.
Mindat. 2020. https://www.mindat.org/
Mineralogical Society of America (MSA) 1997-2020. http://www.minsocam.org/msa/collectors_corner/faq/faqmingen.htm
Mineralogical Society of America (MSA) 2004-2020. http://www.handbookofmineralogy.org/search.html?p=all
Nezat, C.A. Blum, J.D. Yanai, R.D. and Hamburg, S.P. 2007. A sequential extraction to determine the distribution of apatite in granitoid soil mineral pools with application to weathering at the Hubbard Brook Experimental Forest, NH, USA. Applied Geochemistry. 22(11): 2406-2421.
Parikh, S. J. & James, B. R. 2012. Soil: The foundation of agriculture. Nature Education Knowledge. 3(10): 2
Pauling, L. 1929. The principles determining the structure of complex ionic crystals. Journal of the American Chemical Society. 51(4): 1010-1026.
Rutter, N.W. and White, M. 2015. Glaciation. The Canadian Encyclopedia. https://www.thecanadianencyclopedia.ca/en/article/glaciation
Smart, P. and Tovey, N.K. 1981. Electron microscopy of soils and sediments: Examples. Oxford University Press. 400pp.
Soil Classification Working Group (SCWG), 1998. The Canadian system of soil classification. 3rd Edition. Agriculture and Agri-Food Canada. Publication 1646. 187pp.
Spiers, G.A., M.J. Dudas and L.W. Turchenek. 1989a. The chemical and mineralogical composition of soil parent materials in northeast Alberta. Canadian Journal of Soil Science. 69: 721-738.
Spiers, G.A., M.J. Dudas, K Muehlenbachs and L.W. Turchenek. 1989b. Mineralogy and oxygen isotope geochemistry of clays from surficial deposits in the Athabasca Tar Sands area. Canadian Journal of Earth Sciences. 21: 53-60.
Spiers, G.A., Pawluk, S., M.J. Dudas. 1984. Authigenic mineral formation by Solodization. Canadian Journal of Soil Science. 64: 515 532.
St. Arnaud, R.J. 1979. Nature and distribution of secondary soil carbonates within landscapes in relation to soluble Mg++/Ca++ ratios. Canadian Journal of Soil Science. 59: 87-98.
Wang, D. and Anderson, D.W. 2000. Pedogenic carbonate in Chernozemic soils and landscapes of southeastern Saskatchewan. Canadian Journal of Soil Science. 80: 251-261.
Wang, C., Ross G.J., Torrance, J.K., Kodama, H. 1991. The formation of Podzolic B horizons and pedogenic imogolite as influenced by microrelief within a pedon. Geoderma. 50: 63-77.
Warren, C.J. and Dudas, M.J. 1992. Acidification adjacent to an elemental sulfur stockpile: I Mineral weathering. Canadian Journal of Soil Science. 72: 113-126.
Weil, R.R. and Brady, N.C. 2017. The nature and properties of Soils. 15th Edition. Pearson Education. 1086pp.
About the Authors
C. James (Jim) Warren, Ph.D. P.Geo., Land Resource Specialist, Ontario Ministry of Agriculture Food and Rural Affairs, Guelph, Ontario, Canada.

Jim Warren P.Geo. is an environmental soil scientist with a diverse background in research, consulting and teaching. I am a graduate of both the University of Guelph (B.Sc. (Agr), Soil Science), and the University of Alberta (M.Sc. Soil Science; Ph.D. Soil Chemistry). I conducted research in agronomy, geochemistry, and mineralogy at the University of Guelph and University of Waterloo before becoming a private consultant in acid mine drainage. I am currently employed as a Land Resource Specialist conducting soil survey and pedology work with the Ontario Ministry of Agriculture Food and Rural Affairs, and am also an Adjunct Professor with the University of Guelph. I have delivered courses in soil science, soil mineralogy, soil chemistry, geochemistry, and pedology as a sessional lecturer at four universities (Alberta, Guelph, Waterloo, and Toronto). I have authored or co-authored over 100 publications in refereed journals; book chapters, conference proceedings and reports.
How did I get interested in soils? Well, I grew up on a farm where, as a teenager, I spent a lot of time on a tractor doing tillage operations and had a lot of time to think. One of the questions I asked myself during long days in the field was: “What makes plants grow?” With that I spent the next several decades trying to understand soils to answer that question. The quest continutes . . .
Graeme Spiers, Chair of Environmental Monitoring and Professor, School of the Environment, Department of Earth Sciences / Department of Biology, Laurentian University, Sudbury, Ontario, Canada.

Graeme Spiers is currently teaching in the School of the Environment, the Department of Earth Sciences and the Department of Biology, Laurentian University. I obtained a B.Sc. in Earth Science and Botany at the University of Waikato in New Zealand, and M.Sc. and Ph.D. degrees from the University of Alberta, specializing in Pedology, Clay Mineralogy and Chemistry. With commercial laboratory and consulting experience across Canada, I have been associated with research programs at the Universities of Alberta, Guelph, Waterloo, Laurentian and New Brunswick. My research has resulted in numerous technical and scientific publications and presentations in analytical chemistry, aquatic chemistry, environmental geology, ecology, plant biology and soil science. Prior to attending university on a full-time basis, I was a New Zealand dairy farmer for 10 years, earning my undergraduate degree as a long-term hobby between milkings. In Sudbury, I am involved in research initiatives examining the effects of both historical and current anthropogenic metal emissions on soils, rivers and lakes, as well as vegetation within the Sudbury Smelter Footprint. I am actively involved in a variety of research programs across Canada, and actively collaborates with researchers in Russia (Moscow State University and the Russian Academy of Sciences), Australia, New Zealand, China, Peru and South Africa.
A soil composed predominantly of organic matter, in contrast to a mineral soil.
The organic fraction of the soil; includes plant and animal residues at various stages of decomposition, cells and tissues of soil organisms, and substances synthesized by the soil population. It is usually determined on soil that has passed through a 2.0 mm sieve.
The 3.96 billion year old (2.5-4.2 Ga) ancient core of the North American continent. The exposed area is approximately 8,000,000 square kilometers and underlies eastern, central, and northwestern Canada and most of Greenland, extending south into the United States. The igneous and metamorphic rocks are relatively resistant to weathering and erosion but have be exposed to intense and repeated glaciation. Ice during the most recent glaciation, approximately 10,000 years ago, scraped off most of the soil and weathered rock, which was deposited south and southwest of the Shield. Average relief is 30 m, except in northern Labrador and Baffin Island, where it can reach up to 1,500 m.
The most recent glacial episode affecting North America. Also referred to as the Wisconsin Stage or Wisconsin Glacial Episode, it began approximately 75,000 years ago and ended approximately 11,000 years before present.
A class or structural type of silicate in which the SiO4 tetrahedra are lined together in infinite two-dimensional sheets and are condensed with layers of AlO or MgO octahedral in the ratio 2:1 or 1:1. Also called layer silicate minerals.
A phyllosilicate mineral or a mineral that imparts plasticity to clay and hardens upon drying or firing.
The solid-phase surface area (of a soil or porous medium) divided by the solid-particle mass (expressed in m2 g-1), of by the soil-particle volume.
The replacement of one atom by another of similar size in a crystal structure without disrupting or seriously changing the structure. Occurs during the formation of the mineral. When a substituting cation is of a smaller valence than the cation it is replacing, there is a negative charge on the structure. This commonly occurs in phyllosilicates where Al substitutes for Si in tetrahedral sheets and Mg or Fe substitute for Al in octahedral sheets. This substitution usually results in a charge deficit within the structure forming a surface charge site for cation exchange.
(1) A naturally occurring homogeneous solid, inorganically formed, with a definite chemical composition and an ordered atomic arrangement. (2) An economic mineral deposit exploited by mining.
A three-dimensional figure having four triangular sides. It may be visualized as a pyramid with a triangular base. The tetrahedral arrangement can be represented by three balls placed in a triangle in contact with each other and a fourth ball resting on top above the center of the triangle. The normal spacing of the oxygen ions in tetrahedra and other compact structures is the same as the diameter of an oxygen ion, about 0.28 nanometers. The space inside an oxygen tetrahedron is so small that only a very small cation, such as Si4+ or Al3+, can fit inside. Even Al3+ is crowded inside oxygen tetrahedra, and is more frequently found in octahedral spaces.
The electrostatic attraction between oppositely changed ions.
A group of silicate minerals, such as quartz or feldspar, having a structure in which the SiO4 tetrahedra share all four oxygen atoms with neighboring tetrahedra to form a three-dimensional network with a Si:O ratio of 1:2.
Minerals with the same chemical composition but different crystal forms.
The inherent ability of soil to support crop growth, without human intervention.
To infinity.
An octahedral sheet, or a mineral containing such a sheet, that has two-thirds of the octahedral sites filled by trivalent ions such as aluminum or ferric iron.
An octahedral sheet or a mineral containing such a sheet that has all of the sites filled, usually by divalent ions such as magnesium or ferrous iron.
A member of the mica group of minerals with chemical formula K(Li,Al)3(Al,Si,Rb)4O10(F,OH)2
The total amount amount of exchangeable cations that a soil can adsorb; sometimes called "total exchange capacity," "base exchange capacity," or "cation adsorption capacity". It is expressed in centimoles of charge per kilogram of soil or of other adsorbing material (e.g., clay).
A clay that is prone to large volume changes that are directly related to changes in water content. Soils with a high content of expansive minerals can form deep cracks in drier seasons or years; such soils are called vertisols. Montmorillonites expand considerably more than other clays due to the adsorption of water in the interlayer molecular spaces. The amount of expansion depends largely on the exchangeable cation present on the exchange complex. The presence of sodium as the predominant exchangeable cation can result in the clay swelling to several times its original volume.
Polished and striated surfaces that occur along planes of weakness resulting from the movement of one mass of soil against another in soils dominated by swelling clays; commonly observed in Vertisols.
(1) A mineral that has no definite crystalline structure. (2) A mineral that has a definite crystalline structure but appears amorphous because of the small crystallite size. (3) A noncrystalline constituent that either does not fit the definition of allophane or it is uncertain that the constituent meets allophane criteria.
An order of soils in the Canadian system of soil classification having podzolic B horizons (Bh, Bhf, or Bf) in which amorphous combinations of organic matter (dominately fulvic acid), Al, and usually Fe are accumulated. The sola are acid and the B horizons have a high pH-dependent charge. The great groups in the order are Humic Podzol, Ferro-Humic Podzol, and Humo-Ferric Podzol.
An order of soils in the Canadian system of soil classification in which the horizons are developed sufficiently to exclude the soils from the Regosolic order, but lack the degrees or kinds of horizon development specified for soils of the other orders. These soils, which occur under a wide variety of climatic and vegetative conditions, all have Bm or Btj horizons. The great groups Melanic Brunisol, Eutric Brunisol, Sombric Brunisol, and Dystric Brunisol belong to this order.
An order of soils in the Canadian system of soil classification developed under wet conditions and permanent or periodic reduction. These soils have low chromas, or prominent mottling, or both, in some horizons. The great groups Gleysol, Humic Gleysol, and Luvic Glaysol are included in the order.
The upper horizons of a soil in which the parent material has been modified and in which most plant roots are contained. It usually consists of A and B horizons. (plural sola)
