Primary Hemostasis (Platelets)
Primary hemostasis includes the immediate vasospasm or vasoconstriction of the traumatized vessel, followed closely by formation of the platelet plug. Many tiny rents regularly develop in small capillaries. These are normally sealed by platelets. Animals with thrombocytopenia or platelet function defects are usually presented with petechial (pinpoint) hemorrhages visible on superficial surfaces such as oral mucosa and less haired skin. These can occur spontaneously with no history of injury. However, with surgery or trauma, primary hemostatic defects may be manifested as continuous or recurrent oozing from the site, and the bleeding begins at the time of the tissue injury rather than being delayed.
Platelets
In mammals, platelets are anucleate cytoplasmic fragments from the parent megakaryocyte. Megakaryocytes develop from hemopoietic stem cells and have a common progenitor (common myeloid progenitor) with erythrocytes and all leukocytes except lymphocytes (see Chapter 1: Erythrocytes, Fig. 1.1). The path from stem cell to committed megakaryocyte is supported by interleukin 3 (IL-3) and kit ligand. Further development to mature megakaryocytes requires additional cytokines, thrombopoietin being the most important. Thrombopoietin concentrations in the peripheral blood are inversely proportional to the circulating platelet count. The precursor pool of committed megakaryocytes undergoes 15-22 cell divisions over the 3 stages within this pool (Fig. 4.2). Although mitosis (cell division resulting in two daughter cells) ceases at a very early stage, DNA replication continues by a process known as endomitosis, division of chromosomes without accompanying nuclear division. Cytoplasmic granularity becomes more pronounced and abundant through the maturation stages to megakaryocyte. The mature megakaryocyte nucleus usually comprises between 4 (8N) and 32 (64N) times the normal diploid quantity of DNA. The single lobulated nuclear envelope of the mature megakaryocyte contains multiple connected lobules of DNA, each representing one diploid amount (2N) of DNA.
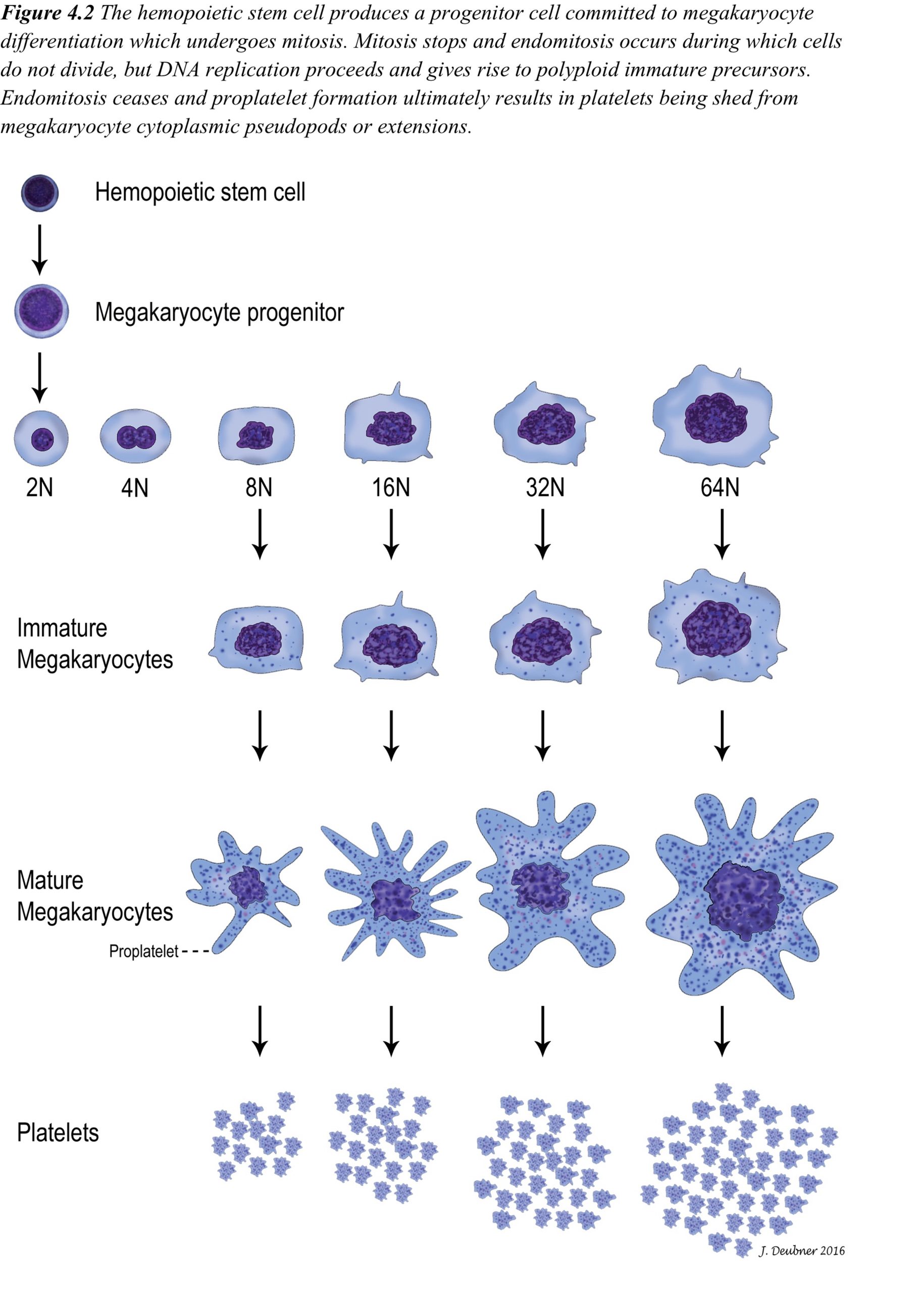
Most megakaryopoiesis (production of megakaryocytes) and thrombopoiesis (production of platelets from megakaryocytes) occurs in the bone marrow but, even under normal circumstances, 7-15% occurs in the spleen and lung where megakaryocytes get trapped in capillary beds as precursors or mature cells. In the bone marrow, megakaryocytes send pseudopods into the sinusoidal lumen and shed their platelets, through pseudopod fragmentation, directly into the circulation. Marrow fibroblasts may be responsible for anchoring immature and mature megakaryocytes to these sites, as well as providing a surface for growth factor expression. Under normal circumstances, 1000-5000 platelets are produced from 1 megakaryocyte but, with increased demand, production can increase 8-fold through the influence of thrombopoietin.
Platelet adhesion
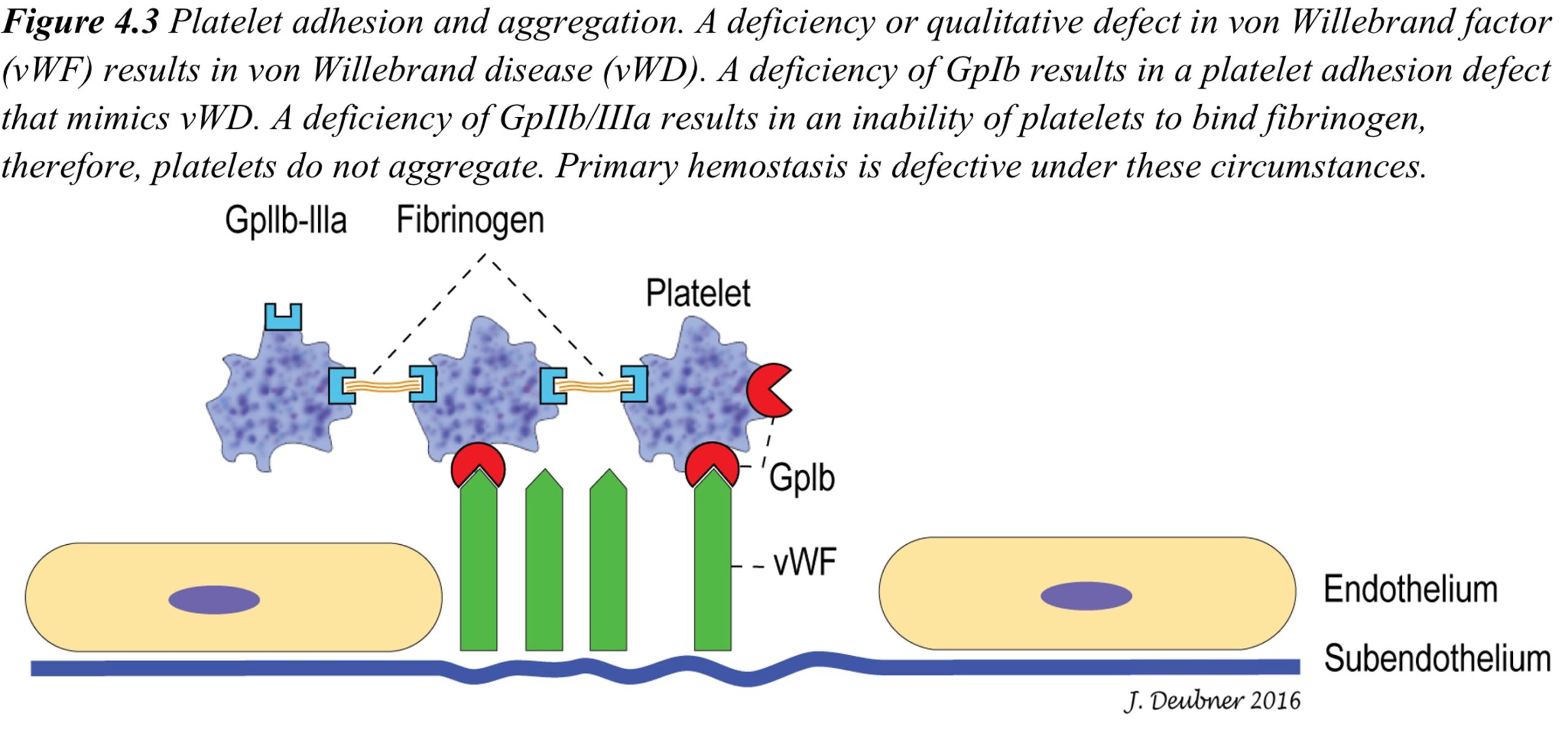
Platelets are highly motile and maintain their disk shape in the circulation unless they become stimulated by exposure to agonists (any substances that elicit a response when bound to a receptor)released from or present on damaged endothelium or exposed subendothelium. Activated platelets change shape from a disk to a sphere and then develop projections or filopods through internal actin filament assembly. Platelets contain a multitude of proteins, growth factors, and coagulation factors, as well as calcium and glycogen. With blood vessel damage, glycoprotein (GP) Ia/IIa receptors on the surface of the platelet bind to exposed collagen in the subendothelium. von Willebrand factor (vWF) strengthens the adhesion by forming additional bonds between collagen and the platelet GP Ib-IX-V receptor, which contains both vWF-binding (GP Ib, see Fig. 4.3) and thrombin-binding sites. This helps explain clinical differences that may be seen with thrombocytopenia compared to certain other disorders of primary hemostasis, such as von Willebrand Disease (vWD), a hereditary condition involving quantitative and/or qualitative deficiencies of vWF. With low shear stress (the frictional forces of blood flow acting on the endothelial surface), GP Ia/IIa-mediated binding of the platelet to the subendothelium may be sufficient to arrest hemorrhage. However, with high shear stress, the strengthened binding of platelets to the subendothelium through the GP Ib-IX-V receptor and vWF may be required to arrest hemorrhage.
Platelet aggregation
Following platelet adhesion, a conformational change in another receptor, GPIIb/IIIa (also known as platelet integrin αIIbβ3), facilitates its ability to bind adhesive proteins, particularly fibrinogen. Interplatelet binding is responsible for platelet aggregation and growth of the primary hemostatic plug. Platelets then become procoagulant through exposure of negatively charged surface phospholipids, binding of tissue factor in the plasma, and release of internal coagulation proteins. Leukocytes are also able to bind to receptors on activated platelets. Repeated cycles of adhesion, aggregation, and release of platelet granule contents perpetuates the growth of the platelet plug and provides a surface for coagulation (fibrin formation) to occur.
In most species, agonist binding to platelet receptors results in the generation of thromboxane A2 from arachidonic acid metabolism. Thromboxane A2 is a potent stimulator of platelet aggregation. Drugs, such as acetylsalicylic acid (ASA), which bind to platelet cyclooxygenase I, inhibit the thromboxane A2 pathway of platelet aggregation and release of granule contents. Platelet aggregation inhibition by ASA is irreversible and therefore lasts for the life of the platelet. Other cyclooxygenase I inhibitors have a reversible effect which lasts for only a few hours. Species variation in the importance of platelet arachidonic acid metabolism is highlighted by the fact that platelet aggregation in cattle is not affected by ASA administration because bovine platelets synthesize little thromboxane.
Decrease in the number of circulating platelets below the reference interval.
Any of a number of conditions (e.g. von Willebrand disease, administration of cyclooxygenase inhibitors such as acetylsalicylic acid) that can lead to defective primary hemostasis despite adequate platelet numbers.
Pinpoint hemorrhage visible on superficial surfaces such as oral mucosa and less-haired skin of animals with thrombocytopenia or platelet function defects.
Polyploid cell found in the bone marrow (also spleen and lung) that produces platelets.
Pluripotential cell that gives rise to all hemopoietic cell lines; also capable of self-renewal.
Can have different hematologic meanings: 1. referring to all hemopoietic cells in the bone marrow except lymphocytes, 2. referring to all bone marrow leukocytes except lymphocytes.
Red blood cell (RBC); an anucleate (in mammalian species) cell containing hemoglobin needed for oxygen transport. Typically shaped like a bi-concave disk.
White blood cell (WBC); includes neutrophils, eosinophils, basophils, monocytes, lymphocytes, mast cells.
Mononuclear, non-phagocytic leukocyte responsible for humoral (B lymphocyte) and cell-mediated (T lymphocyte) immune responses.
Growth factor that maintains platelet mass.
Process by which DNA replication occurs without cell division; e.g. within megakaryocytes.
Part of hemopoiesis dealing with the production of megakaryocytes.
Part of hemopoiesis dealing with the production of platelets from stem cells to megakaryocytes to circulating platelets.
Organ responsible for production of hemopoietic cells; found in the medullary cavity, especially the ends of long bones (e.g. femur) and flat bones (e.g. the pelvis, sternum.)
Negatively charged surface that is exposed following vascular injury.
Erythrocyte cytoskeletal protein.
Protein important in binding platelets to subendothelial collagen.
Platelet surface protein that binds to von Willebrand factor and thrombin
Central protein of coagulation; not only converts fibrinogen to fibrin, but also plays important roles in amplification and fibrinolysis.
Disorder of primary hemostasis due to qualitative or quantitative defects in von Willebrand factor. The most common inherited bleeding disorder in dogs.
Platelet surface protein that binds fibrinogen
Type of lipid used to form cell membranes.
Protein released from damaged tissue important in initiating coagulation.
The end product of coagulation, produced by the conversion of fibrinogen.
Chemical mediator that stimulates platelet aggregation in most species (not cattle).
